Servicio de Neurología UNIVERSIDAD AUTÓNOMA DE MADRID FACULTAD DE MEDICINA Ana Frank García Hacia el diagnóstico genómico personalizado temprano de la enfermedad de Alzheimer 14 Enero 2012 Diplomatura de Postgrado de Medicina del Envejecimiento, Xª Edición. • Necesidad de diagnóstico precoz DCL • Diferenciar EA de otras demencias – Avance en técnicas de imagen (RM, PET) – Biomarcadores (LCR y sangre) • Futuro Tratamiento modificador de evolución
Transcript
Servicio de NeurologíaUNIVERSIDAD AUTÓNOMA DE MADRID
FACULTAD DE MEDICINA
Ana Frank García
Hacia el diagnóstico genómicopersonalizado temprano de la enfermedad
de Alzheimer
14 Enero 2012
Diplomatura de Postgrado de Medicina del Envejecimiento, Xª Edición.
• Necesidad de diagnóstico precoz DCL• Diferenciar EA de otras demencias
– Avance en técnicas de imagen (RM, PET)– Biomarcadores (LCR y sangre)
• Futuro Tratamiento modificador de evolución
3
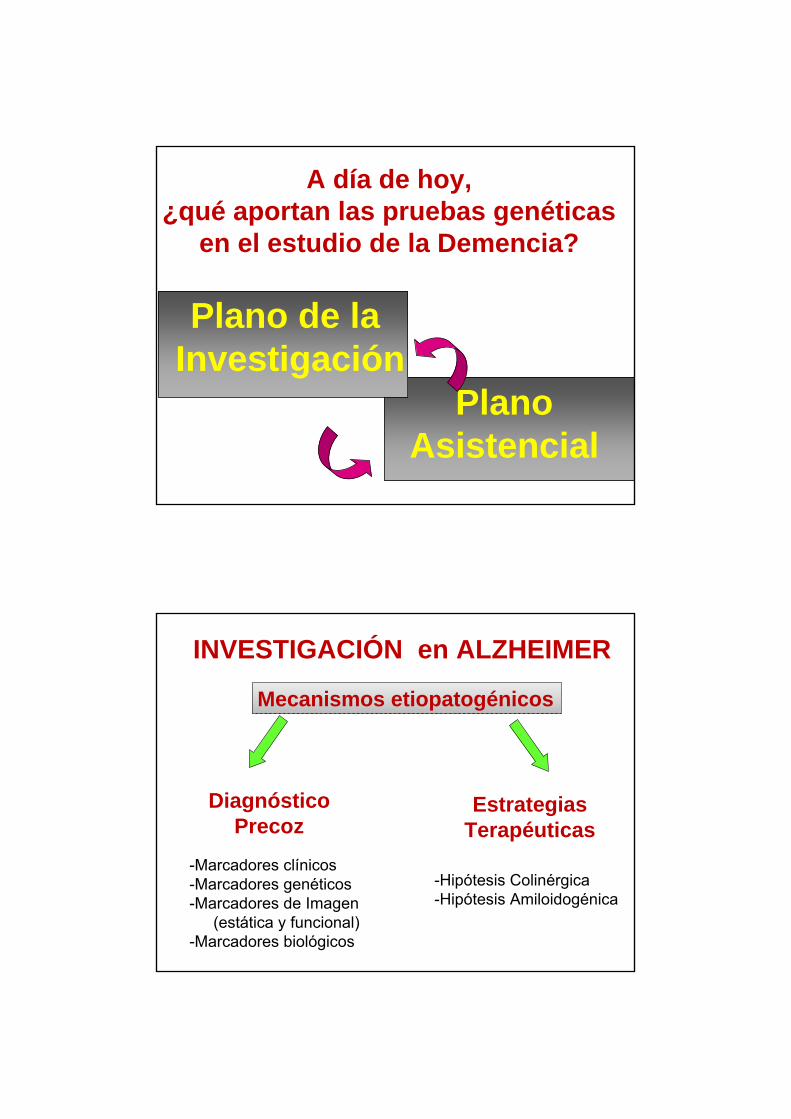
Diagnóstico precoz
Tratamiento modificador de evolución
Biomarcadores
INVESTIGACIÓN EN DEMENCIAS (EA)
Contenidos• Conceptos básicos
• Fisiopatología de la E. Alzheimer
• Mutaciones y Polimorfismos
• Estudios genéticos en demencias
• Debate
• Conclusiones
Gen• Parte “elocuente” del cromosoma• Cada gen codifica para una proteína
• Anomalías Mutaciones• Variaciones Polimorfismos
Cromosoma• Localizados en el núcleo de las células• Anomalías P.e. Trisomías (S. Down)• Cada Cr posee muchos genes
Especie humana23 pares de cromosomas
1961Premio Nobel
Watson & Crick
Modelo DNA
Nucleótido
A - F - A - F - A - F - A - F
B B BBTriplete de bases
Cada codóncodifica para 1 aminoácido
• Azúcar (desoxirribosa)• Fosfato• Base
•Púrica (Adenina, Guanina)
• Pirimídinica (Timina, Citosina)
Constitución del DNA A - F - A - F - A - F - A - F
B B BB
A - F - A - F - A - F - A - F
B B BB
Codón
Cariotipo humano
Aspectos genéticos de las enfermedades
Mutación Proteínaanómala
Polimorfismo¿Factores
ambientales?
Función alterada
Enfermedad
Gen
Cromosoma
Proteínanormal Función Fisiológica Salud
Proteínadisfuncional Susceptibilidad
+
Genoma Humano23 pares de cromosomas
Identificar cromosoma
Identificar región
Identificar Gen
Identificar Mutación
Libro de la Vida23 pares de volúmenes
Identificar el volumen
Identificar el capítulo
Identificar la página
Identificar el errortipográfico
Detección de mutaciones puntuleso polimorfismos
ETAPAS CRONOLÓGICAS DE LA GENÉTICA
1865 (1900) – 1940: Genética de la transmisión
1940 – 1960: Naturaleza y propiedades del material hereditario
1960 – 1975: Mecanismos de acción génica: Expresión (código, transcripción, traducción) y regulación de los genes. Desarrollo
1975 – 1985: Nueva Genética, basada en la tecnología de los ácidos nucleicos (fragmentación, hibridación, secuenciación, amplificación)
Activación microglial y astrocítica(citokinas, factores del complemento) Daño neurítico y sináptico progresivo
SSÍÍNTOMASNTOMAS
DAÑO NEURONAL
MUERTENEURONAL
ALTERACIÓNNEUROTRANSMISIÓN
+
RECOMENDACIONES DEL NIAAA (“NATIONAL INSTITUTE ON AGING &
ALZHEIMER´S ASSOCIATION” )
• Clasificación de los biomarcadores en 2 grupos– I : de acumulo de β-amiloide (PET-PIB, descenso en LCR)– II: de daño neuronal (tau/ P-tau en LCR, PET-FG, atrofia
hipocampo-temporal IRM, otros…)
• Patocronia: I II
• 3 síndromes, cronológicamente– EA preclínica: Sólo biomarcadores (BBMM)– DCL por EA (EA “prodrómica”): Clínica ± BBMM– Demencia por EA: Clínica “matizada” por BBMM
Toward defining the preclinical stages of Alzheimer’s disease
Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease
Sustitución de nomenclatura clásica de EA “posible, probable y definida”
por nuevo léxico: EA Preclínica y EA Prodrómica• EA PRECLÍNICA
– Asintomática (“estado de riesgo”): biomarcadores + (Fisiopatogénicos) en paciente asintomático
– Presintomática: Marcadores genéticos AD + (mutación PSN I, II o APP) en paciente asintomático
• EA PRODRÓMICA (“estadio predemencia” en la EA)– Déficit mnésico de perfil hipocámpico, +/- otros déficit cognitivos , Y– Biomarcadores positivos (Fisiopatogénicos), Y– No interferencia significtativa en actividades instrumentales de la vida diaria (no
demencia DSM-IV)– ~DCL amnésico (uni o multidomnio) con 2 condiciones:
• DETERIORO COGNITIVO LIGERO:– Dx de EXCLUSION: situación que no cumple los criterios de EA prodrómica
Déficit mnésico HIPOCAPMPICOBIOMARCADORES +
EA PRODRÓMICA(Dubois et al)
• Imprescindible déficit mnésico hipocámpico
• Biomarcadores– Al menos uno Positivo
• Genética– Mutaciones AD
DCL por EA(NIAAA)
• Se acepta déficit en otros dominios ≠ de memoria
• Biomarcadores– Asistencia no necesarios– Investigación Necesarios
• Genética– Mutaciones AD o ApoE 4
Nuevo Léxico para E. Alzheimer
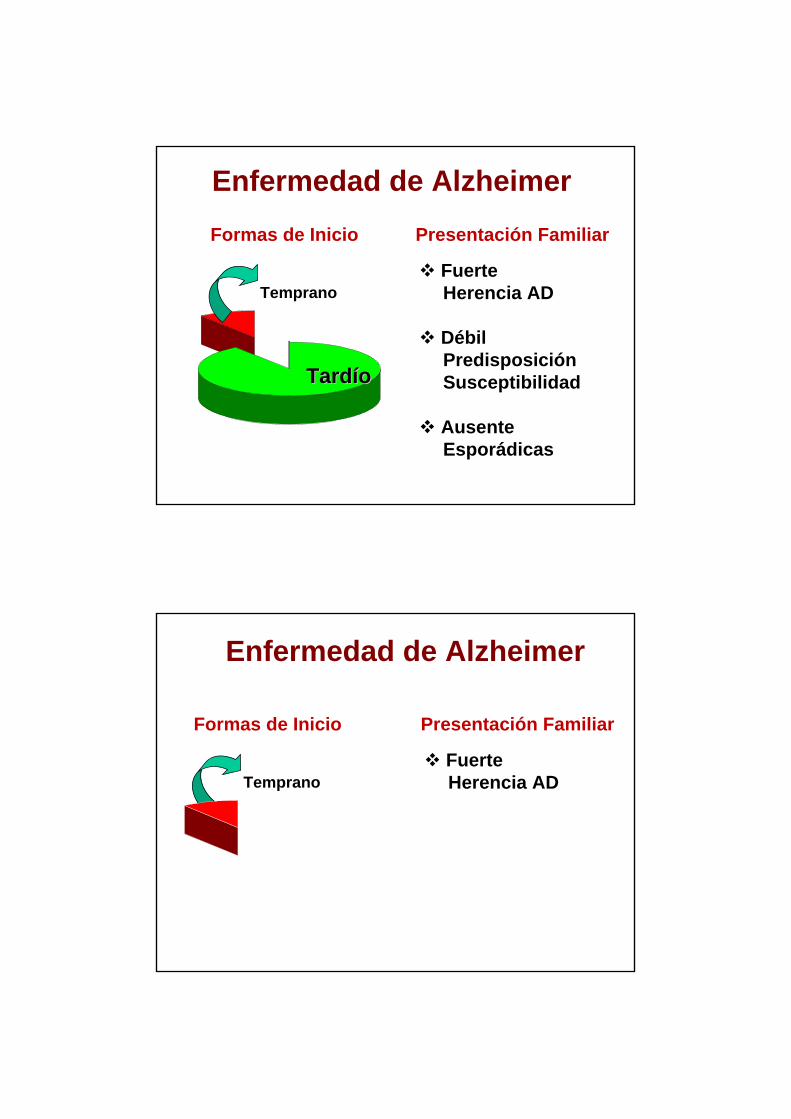
Formas de Inicio
Temprano
TardTardííoo
Presentación Familiar
Fuerte Herencia AD
DébilPredisposiciónSusceptibilidad
Ausente Esporádicas
Enfermedad de Alzheimer
Formas de Inicio
Temprano
Presentación Familiar
Fuerte Herencia AD
Enfermedad de Alzheimer
- Gen de Presenilina 2 (PS-2)(Cromosoma 1)
- Gen de Presenilina 1 (PS-1)(Cromosoma 14)
-Gen de Proteína Precursora de Amiloide (APP)
(Cromosoma 21)
Mutaciones que explican casos de EA familiar de comienzo temprano
(Representan solamente 2-10% de todos los casos)
Mutaciones causantes de EAPS1 > PS2 > PPAGran impacto cara desentrañar fisiopatología
Hipótesis amiloidogénicaDesarrollo de terapias “anti-amiloide”
Etiología de la enfermedad de Alzheimer
Compleja95-99%
Agregación familiar
Genética1-5%
Herencia autosómicadominante
< 5% de todos los casos de enfermedad
Escaso impacto como
factor de riesgo
Mutaciones causantes de E.A.Formas de Inicio
Temprano
Tardío
Escaso impacto como factor de riesgo
PSEN-1 > PSEN-2 > PPAGran impacto cara desentrañar fisiopatologíaHipótesis amiloidogénicaDesarrollo de terapias “anti-amiloide”< 5% de todos los casos de enfermedad
En qué casos habría que realizar estudios de mutaciones?
Investigación
Asistencia clínica
Hª familiar de demencia AD
Casos inicio temprano
Aplicaciones clínicas en DCL (EA Prodrómica)
Consejo Genético factible
Explicación del riesgo
Posibilidad de organización vital
¿Otras?
Enfermedad de Alzheimer
Formas de Inicio
TardTardííoo
Presentación Familiar
DébilPredisposiciónSusceptibilidad
Ausente Esporádicas
TardTardííoo
- Gen de APOE (Alelo ε4)(Cromosoma 19)
- ¿Otros?
Polimorfismos implicados en casos de EA familiar y esporádicos de comienzo tardío(Representan > 90% de todos los casos)
APOE -/4APOE-/-
APOE E. ALZHEIMER INICIO TARDÍO
APOE 4/4
APOE y diagnóstico de E. Alzheimer
Poseer alelo ε4 da mas posibilidades de que una persona con clínica de demencia tenga EA en vez de otras demencias.
En ausencia de contexto no puede descartar ni afirmar el diagnóstico.
APOE4 no es condición ni necesaria ni suficiente para el dx de E. Alzheimer.
Factores de riesgo genético– Inicio temprano: <60 años ~ 5% casos
• La mitad son casos familiares• Herencia AD (mutaciones APP; PSEN1; PSEN2)
– Participan varios genes con efecto menor– Papel de factores ambientales
• Gen APOE• Año 2007 Tecnologías de genotipado de alto rendimiento
(high-throughput genotyping). – Genome-Wide Association studies (GWAs) Estudios de
asociación del genoma entero para la enfermedad de Alzheimer.
Núria Setó-Salvia y Jordi Clarimón. Genética y enfermedad de Alzheimer [en línea]. La Circunvalación del hipocampo, junio 2010 [Consulta: 25 junio 2011]. Disponible en: http://www.hipocampo.org/originales/original0008.asp
Genes asociados con los fenotipos de EA
EA deinicio temprano
EA deinicio tardío
APPPS1 PS2
(> 100 mutaciones con herencia AD)
APOE (alelo 4) (OR: 33.1)
Efecto patogEfecto patogéénico mediante el nico mediante el acacúúmulomulo de de ppééptido ptido ββ--amiloideamiloide en el cerebroen el cerebro
Eje amiloidogénico Terapias anti-amiloide
Single-nucleotide polymorphisms (SNP)• Variación de la secuencia de DNA
entre individuos de la misma especie o en 2 cromosomas pareados
• Es el resultado de que 1 nucleótido simple (A-G-C-T) cambia.
• Ejemplo: en estos 2 fragmentos de DNA
• AAGCCTA• AAGCTTA
únicamente hay 1 cambio en un solonucleótido.
ESTUDIOS DE ASOCIACIÓN GENÉTICA
Pacientes
Paridad (odds) 6/4 = 1.5
Controles
Paridad (odds) 2/8 = 0.25
Riesgo (odds ratio)
6 / 4
2 / 8= 6
• Sujetos de estudio: Caso-Control• Marcadores: Muy abundantes (SNPs)• Resultado: Odds Ratio
Estudios genéticos en demencias
• Escasos genotipos con baja prevalencia y alta expresividad clínica
• Herencia AD (Mutaciones)– E. Alzheimer– Otras Demencias (DFT; Parkinson-demencia; E.
Huntington; ECJ familiar; CADASIL, etc)
• Abundantes genotipos con alta prevalencia y baja expresividad clínica
• Herencia compleja, poligénica(Fact. riesgo / susceptibilidad) Polimorfismos– E. Alzheimer
Genes asociados con la enfermedad de Alzheimer compleja
1993: APOE
1995:a1-antiquimotripsinaReceptor de VLDLPresenilina-1
1996:α-sinucleína
1997: Transportador de serotoninaProteína rel con LDLR (LRP)ButirilcolinesterasaTransferrina
1998:Promotor de APOE (APOE-p) Neurotrofina-3Bleomicinahidrolasaα2-Macroglobulina(A2M)Receptores de serotoninaFE65
2000: IL-1a
2002:Oestrogenreceptor β
1999:ACE MieloperoxidasaDihidrolipoilsucciniltransferasaCatepsina D (*)HLA-A2N-acetil transferasaLipoproteína lipasaIL-6 (*)NOS3HLA-DRB1Receptor de
estrógenosTau
Polimorfismos• APOE• CLU, CR1, PICALM
- Gen de APOE (Alelo ε4)(Cromosoma 19)
- ¿Otros genes?
Etiología de la enfermedad de Alzheimer
Compleja95-99%
Agregación familiar
Genética1-5%
Herencia autosómicadominante
2009
Alzheimer’s disease beyond APOEMichael A van Es & Leonard H van den Berg Nature Genetics 2009; 41: 1047-8
GWAs 3 nuevos loci de susceptibilidad para E. Alzheimer. CLU, PICALM y CR1
involucrados en aclaramiento cerebral de β-amiloide.
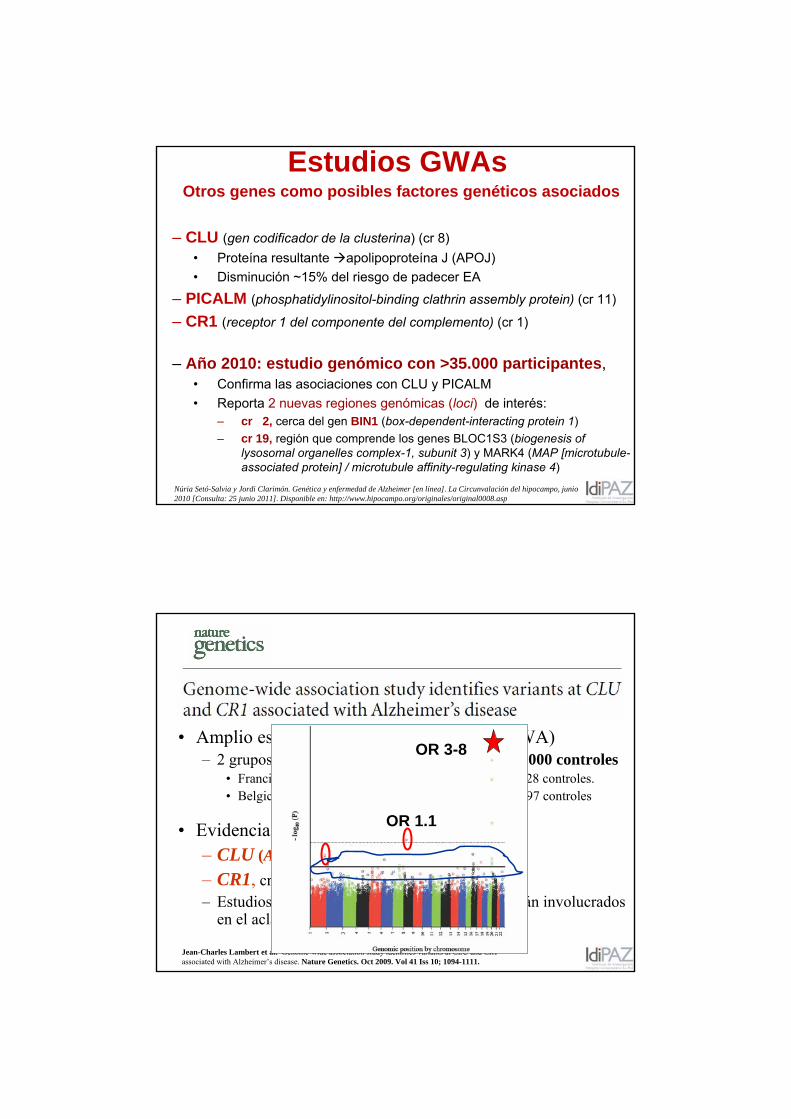
Estudios GWAsOtros genes como posibles factores genéticos asociados
– CLU (gen codificador de la clusterina) (cr 8) • Proteína resultante apolipoproteína J (APOJ)• Disminución ~15% del riesgo de padecer EA
– CR1 (receptor 1 del componente del complemento) (cr 1)
– Año 2010: estudio genómico con >35.000 participantes, • Confirma las asociaciones con CLU y PICALM• Reporta 2 nuevas regiones genómicas (loci) de interés:
– cr 2, cerca del gen BIN1 (box-dependent-interacting protein 1)– cr 19, región que comprende los genes BLOC1S3 (biogenesis of
Núria Setó-Salvia y Jordi Clarimón. Genética y enfermedad de Alzheimer [en línea]. La Circunvalación del hipocampo, junio 2010 [Consulta: 25 junio 2011]. Disponible en: http://www.hipocampo.org/originales/original0008.asp
Jean-Charles Lambert et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nature Genetics. Oct 2009. Vol 41 Iss 10; 1094-1111.
• Amplio estudio de asociación genómica (GWA) – 2 grupos de muestras: total de ~ 6000 EA + > 8000 controles
• Francia: 2.032 EA + 5.328 controles. • Belgica, Finlandia, Italia y España: 3.978 EA + 3.297 controles
• Evidencia de asociación genómica en 2 loci:– CLU (APOJ), cromosoma 8 – CR1, cromosoma 1 – Estudios previos sugieren que CLU and CR1 están involucrados
en el aclaramiento de péptido Aβ
OR 1.1
OR 3-8
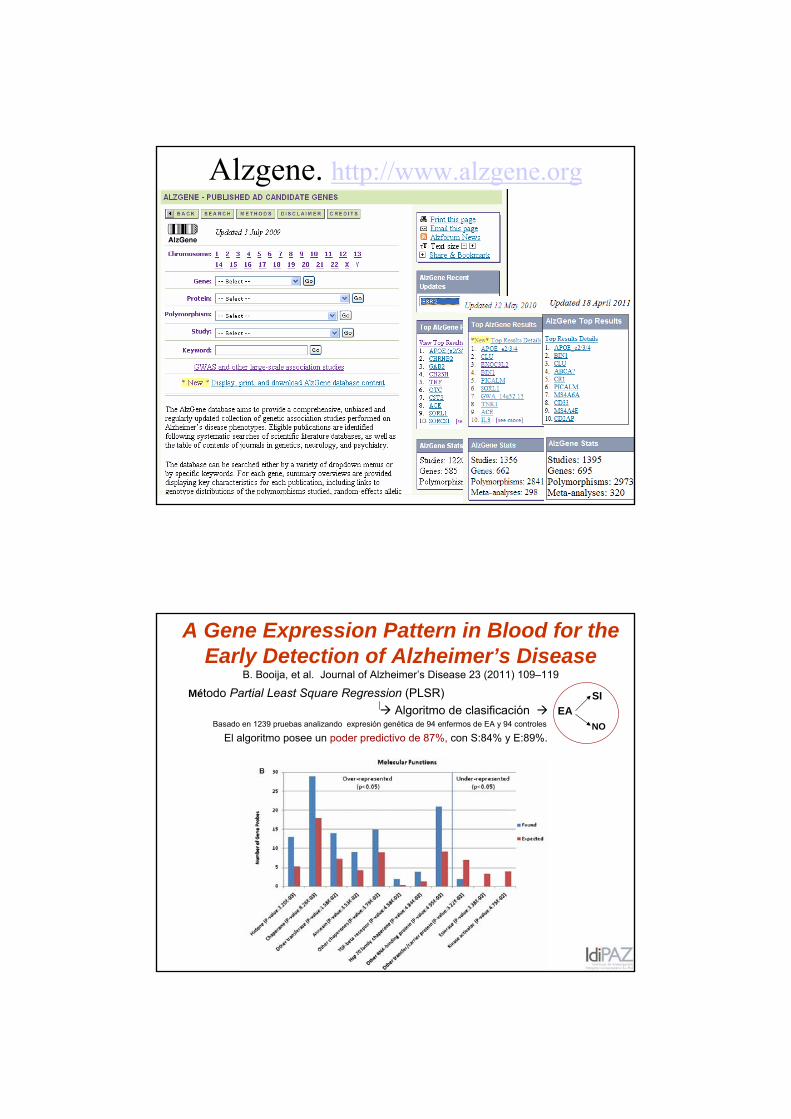
Alzgene. http://www.alzgene.org
A Gene Expression Pattern in Blood for the Early Detection of Alzheimer’s Disease
B. Booija, et al. Journal of Alzheimer’s Disease 23 (2011) 109–119
Método Partial Least Square Regression (PLSR) Algoritmo de clasificación
Basado en 1239 pruebas analizando expresión genética de 94 enfermos de EA y 94 controles
El algoritmo posee un poder predictivo de 87%, con S:84% y E:89%.
EASI
NO
A Novel Blood Test for the Early Detection of Alzheimer’sDiseasePhil. D. Ryea,et al.Journal of Alzheimer’s Disease 23 (2011) 121–129
• A disease classification algorithm was developed on samples from 208 individuals (AD = 103; controls = 105) and was validated in two steps using an independent initial test set (n = 74; AD= 32; controls = 42) and another second test set (n = 130; AD= 68; controls = 62).
• In the initial analysis, diagnostic accuracy was 71.6±10.3%, with sensitivity 71.9±15.6% and specificity 71.4±13.7%. Essentially the same level of agreement was achieved in the two independent test sets.
• High agreement (24/30; 80%) between algorithm prediction and subjects with available cerebrospinal fluid biomarker was found.
• Assuming a clinical accuracy of 80%, calculations indicate that the agreement with underlying true pathology is in the range 85%-90%.
• These findings suggest that the gene expression blood test can aid in the diagnosis of mild to moderate AD, but further studies are needed to confirm these findings.
Reorgnización neuronalCascada amiloide
Estrés OxidativopTAU
Inflamación
Estrés OxidativoEfectos en la Transcripción
Genes con efectos
sistémicos
Genes con efectos locales
Composición de ADtect®96-gene signature
ADtect® analiza• Genes implicados en el ciclo y metabolismo celulares• Amplio abanico de vías relacionadas con la EA
FamiliaresE. Creutzfeldt-Jakob familiarS. Gerstmann-Sträussler-ScheinkerInsomnio Fatal FamiliarEnfermedades por priones atípicos
Adquiridas (transmisibles)KuruE. Creutzfeldt-Jakob yatrogénicaNueva variante de E. Creutzfeldt-Jakob
Herencia ADPenetrancia variabledependiente del tipo de mutación en gen PRNP(Cr20p).
Mayoría mutacioneslocalizadas en codón 200.
Gen Huntington (Cr 4)
• Indicación:– Sospecha de E. Huntington– Consejo genético en familiar
asintomático
Se debe seguir el protocolo de actuación establecido
FACTORES GENÉTICOS INVOLUCRADOS EN ENFERMEDAD
DE ALZHEIMERAPP
APOE-4
PS-1 PS-2
DesconocidosDesconocidos
¿A QUIÉN?
¿CUÁNDO?
¿DÓNDE?
Estudios genéticos
Principios Bioéticos• Autonomía• Beneficiencia• No maleficiencia• Justicia• Integridad• Dignidad• Vulnerabilidad
• Consentimiento informado• Confidencialidad• Validez científica• Selección justa• Balance riesgo/beneficio• Revisión independiente• Derecho a no saber
Proporcionar la información del estudio genético
Consentimiento informado•Relación médico-paciente-familia•Información veraz y adaptada al nivel cultural
Apoyo psicológico• Estrategias para afrontar consecuencias
Confidencialidad
Bertram y Tanzi. J. Clin. Invest. 115:1449-1457 (2005).
Genes y ambiente participan en la etiopatogenia de la EA
N. L. Pedersen. Reaching the Limits of Genome-wide Significance in Alzheimer Disease. Back to theEnvironment. JAMA, May 12, 2010—Vol 303, No. 18
• The value of continued attempts to find genetic effects of diminishing importance remains uncertain.• Important questions are whether these small effect sizes have any value in understanding disease
pathogenesis and what truly are the clinical implications of this line of study.• Alzheimer disease is one of the most heritable common, complex disorders, with a heritability of 60% to
80%, and is one of the few diseases for which a single susceptibility gene, apolipoprotein E (APOE), gives rise to a substantial risk.
• Might it be that AD is not just polygenic but is the result of risk alleles in a cluster of genes (most oftenincluding APOE) where some constellations of risk alleles are important in some individuals while othercombinations are important in other individuals?
• Or might differing combinations of risk alleles and environmental triggers (manifested as gene-environment interactions) be thwarting the ability to predict risk?
• Clearly, researchers need to pay much more attention to environmental risk and protective factors• Environmental risk factors may accumulate with age or act as triggers of a disease process in a
susceptible brain.• What does this newly found genetic knowledge tell clinicians? It is a fresh reminder that family history is
very important, even for late-onset disease that was once thought to be sporadic.• Lessons from increasing numbers of epidemiological studies with prospective information indicate
that changes in midlife behavior, particularly those that are also conducive to cardiovascular health, can reduce risk of dementia or at least postpone onset.
El creciente nº de estudios epidemiológicos prospectivos nos enseña que los hábitos en edades medias de la
vida -en particular los concernientes a la salud cardiovascular- pueden ↓↓ el
riesgo de demencia o, al menos, retrasar su inicio.
• En el plano de investigación los estudios genéticos (mutaciones y polimorfismos) están siempre indicados tanto en pacientes como en familiares asintomáticos.
• En el plano asistencial solo están indicados para contribuir al diagnóstico etiológico de pacientes con sospecha de mutaciones específicas y para el consejo genético de familiares asintomáticos.
• Los genotipos de susceptibilidad (APOE) no contribuyen a establecer el diagnóstico, por tanto NO están indicados.
Conclusiones
• Es preciso que los estudios genétios se lleven a cabo por un equipo multidisciplinario especializado– que garantice el cumplimiento de todos los
requerimientos éticos y – que pueda proporcionar un adecuado
asesoramiento psicológico.
• Los estudios genéticos en demencias siempre deberán llevarse a cabo siguiendo el procedimiento acorde a los principios bioéticos.