

Provided by
Tumores Renales. Nefroblastoma(Tumor de Wilms)
Lead contributors:
Jaume Mora, MDHospital Saint Joan de Deu
Barcelona, España
A. Introducción
El año 1877 el Dr. Thomas Jessop (1837–1903) realizó con éxito la
primera nefrectomía a un niño de 2 años con hematuria debida a un gran tumor
de su riñón izquierdo. En 1899 el también cirujano Max Wilms (1867–1918)
describió por primera vez 7 niños afectos de un tumor que él denominó
nefroblastoma, lo que ha venido a conocerse posteriormente como el tumor de
Wilms. Sabemos ahora que el nefroblastoma, o tumor de Wilms, es la neoplasia
del riñón más frecuente durante el desarrollo (85%), aunque no la única. El
tumor de Wilms era una neoplasia inexorablemente fatal a principios del
siglo XX pero con el desarrollo de las técnicas quirúrgicas y, sobretodo, el
descubrimiento de su gran radiosensibilidad y la introducción de la
quimioterapia, cambiaron drásticamente las posibilidades de curación. Hoy día,
con supervivencias superiores al 90%, la prioridad en el estudio del tumor de
Wilms es la disminución de las secuelas relacionadas con el tratamiento
manteniendo el elevado índice de curación.

Page 2 of 30
A.1 Epidemiología
El tumor de Wilms (TW) afecta aproximadamente a de cada 10,000 niños
menores de 15 años, lo que supone una incidencia anual de 7,6 casos por
millón de individuos por debajo de los 15 años de edad o unos 600 nuevos
casos en USA y 30-40 en España. El tumor de Wilms representa un 6% de todos
los tumores en esta franja de edad, o la cuarta neoplasia del desarrollo después
de la leucemia, los tumores del sistema nervioso central tomados en su
conjunto y el neuroblastoma. Su incidencia ha permanecido estable durante los
últimos 30 años, y de hecho se utiliza su estabilidad para medir los cambios de
frecuencia en otras neoplasias infantiles, que si parece que cambian. El tumor
habitualmente aparece en un solo riñón antes de los 5 años de edad, con la
misma frecuencia para los dos géneros. La media de edad de presentación en
los casos unilaterales es de 43 meses y de 31 para los casos bilaterales. Su
presentación antes del año de vida es rara y debe sugerir condiciones genéticas
predisponentes (ver abajo) y después de la pubertad, excepcional. Por la edad
de presentación, sus características histológicas y su recapitulación de los
procesos embriológicos que describimos a continuación, hacen del tumor de
Wilms uno de los ejemplos más emblemáticos de los llamados tumores
embrionarios propios del desarrollo.
A.2 Embriología
El TW es un tumor que recapitula de manera asombrosa el complejo
proceso de la formación del riñón. El metanefros, o riñón definitivo, es el
principal órgano de filtración del organismo y su desarrollo embriológico
requiere de la interacción de varios tejidos que a su vez padecen múltiples
transformaciones morfogenéticas. La inducción de la formación del metanefros
ocurre por la interacción recíproca entre el componente epitelial, o ducto
mesonéfrico (o Wolffiano), y el mesénquima metanéfrico (o blastema).

Page 3 of 30
Las nefronas se forman continuamente en el córtex del riñón en desarrollo
a partir de células precursoras del mesénquima (blastema) metanéfrico no
inducido. Estas células del blastema metanéfrico se encuentran en el córtex del
riñón en desarrollo muy cerca de la cápsula renal y presentan características
propias de una célula madre (stem cell): sus descendientes pueden mantenerse
indiferenciadas o diferenciar a cualquiera de los distintos tipos celulares que
conforman el riñón maduro, por lo tanto son células con capacidad
multipotencial. La inducción de la formación de nefronas debe estar finamente
ajustada para evitar la masiva transformación de la población del mesénquima
blastémico en nefronas. A su vez, los defectos en la inducción de la
formación de nefronas lleva a la persistencia de restos nefrogénicos, a los que
se considera como las lesiones precursoras del tumor de Wilms. Los restos
nefrogénicos ocurren de manera infrecuente en riñones normales, pero en
pacientes con TW más de un tercio de los riñones intervenidos quirúrgicamente
contienen restos nefrogénicos.
Estos restos se clasifican como perilobares o intralobares. Los perilobares
son habitualmente multifocales y consisten histológicamente de blastema y
túbulos. Los intralobares son habitualmente unifocales y están compuestos
habitualmente de un estroma predominante con presencia acompañante de
blastema y túbulos. Los perilobares se asocian a síndromes de hipercrecimiento
como la hemihipertrofia y el síndrome de Beckwith-Wiedemann, mientras que
los intralobares se encuentran habitualmente en los síndromes WAGR (Wilms
tumor-aniridia-genital anomaly-mental retardation) y Denys-Drash (DDS). Ambos
tipos de restos, y los síndromes asociados, están causados por alteraciones en
vías moleculares características que se describen a continuación y se sintetizan
en la Tabla 1.

Page 4 of 30
Tabla 1
Mutaciones de WT1 LOIIgF2
Restos nefrogénicos Intralobares Perilobares
Localización Profunda Periférica
Nefrogénesis Alteración precoz Alteración tardía
Diagnóstico Precoz (16 meses) Tardío (36meses)
Peso al nacer normal/bajo alto
Genéticaacompañante
Mutacions de B-catenina
Hipermetilaciónde H19
Sindromesasociados WAGR y DDS BWS y
hemihipertrofia
Histología Tumoresrabdomiomatosos
Tumoresblastematosos
WT1inmunohistoquímica ausente/citoplasmática Nuclear
Respuesta a laquimioterapia Baja Buena
Tratamiento CirugíaCirugía +
quimioterapia+/-Radioterapia
Riesgo Nuevos tumores adquisición deresistencias
B. Etiopatogenia
B.1 Genética y biología molecular
La predisposición a desarrollar nefroblastoma es bien conocida en
algunos raros síndromes hereditarios por lo que rápidamente se sospechó que
existían factores genéticos involucrados en la patogenia del TW. Un primer
grupo de síndromes están caracterizados por anomalías en el desarrollo

Page 5 of 30
genitourinario (como el WAGR y DDS) y el otro por sobrecrecimiento (como el
Beckwith-Wiedemann). En el primer grupo se ha descrito el gen WT1 y en el
segundo WT2 como los genes predisponentes primarios. No obstante, en los
últimos años se ha descubierto que múltiples genes contribuyen a la formación
del TW, unos como alteraciones primarias que predisponen al desarrollo del
tumor y otros como eventos secundarios asociados a la progresión maligna.
Ya en la descripción original del modelo de cáncer de A. Knudson basado
en la doble mutación (germinal y somática) se utilizó el retinoblastoma, el
neuroblastoma y el TW como paradigmas. En ese modelo, basado en estudios
epidemiológicos, las formas tumorales bilaterales o multicéntricas
corresponden a casos hereditarios, que reciben la primera mutación de las
células germinales. Hoy sabemos, sin embargo, que sólo un 10% de los TW son
bilaterales, y sólo una fracción de éstos se asocian a síndromes genéticos
predisponentes y los casos de TW familiar son extraordinariamente raros
(estimado en <1% de los casos diagnosticados de TW).
B.1.1 WT1
El análisis citogenético de los individuos afectos de WAGR mostró
deleciones en el cromosoma 11p13, una región que posteriormente se
demostró que alberga el locus contiguo de los genes PAX6, uno de los genes
que causan aniridia, y WT1, uno de los genes primarios que predisponen al
desarrollo de TW (Figura 1). WT1 codifica un factor de transcripción que es
clave para el desarrollo normal del riñón y las gónadas.

Page 6 of 30
Figura 1*
*Silván, Ana María. Aniridia and Wilms Tumor, Oncopedia #289. Released on Oncopedia: 05/11/2009.URL: https://www.cure4kids.org/ums/oncopedia/case_detail/?id=289
WT1 es necesario para la inducción del mesénquima metanéfrico y tiene
una función clave en la formación y diferenciación final de nefronas y
podocitos. Por ello, la pérdida de la función de WT1 durante el desarrollo del
riñón causa un bloqueo en la diferenciación de los precursores nefrogénicos
dejándolos en un estado de célula multipotencial.
La adquisición añadida de mutaciones en otros genes que estimulan el
crecimiento sería aún necesaria para el desarrollo final del TW.
Así, en la mayoría de casos con mutaciones de WT1 se encuentran también
mutaciones en el gen β-catenina, una proteína clave en la vía de WNT. Las
mutaciones de β-catenina afectan habitualmente a la región que codifica para la
señal de degradación por lo que suelen conllevar una acumulación patológica
de la proteína en el núcleo. Dicha acumulación conlleva la activación
transcripcional aberrante y muy probablemente a la promoción de la
proliferación de las células precursoras y con ello al desarrollo del TW.

Page 7 of 30
Muy recientemente se ha descubierto WTX, un nuevo gen mutado en al
30% de los TW. WTX es una proteína que se asocia con β-catenina y promueve
su degradación. Una vez más, mutaciones de genes que causan una
acumulación de β-catenina promueven la proliferación de células precursoras
tumorales favoreciendo el desarrollode TW. WTX es el primer gen supresor de
tumores que se aísla en el cromosoma X, del cual tanto los hombres como las
mujeres sólo tienen una copia funcional del gen, así una sola mutación es
necesaria para su inactivación. Curiosamente la mutación germinal de WTX
predispone a una displasia ósea hiperostótica pero no predispone a la
formación de tumores ni a TW, lo que muestra cómo las alteraciones en la vía
de WNT deben darse de manera coordinada en la célula precursora y durante la
nefrogénesis para
causar TW.
El síndrome Denys-Drash (DDS) está causado por una mutación en la
región de WT1 que se une al DNA lo que da lugar a una proteína con efecto
dominante negativo. Clínicamente DDS se caracteriza por la presencia de
pseudohermafroditismo, glomerulopatía, fallo renal, y una probabilidad de
desarrollar un TW del 95%. Las mutaciones homocigotas de WT1 se encuentran
en alrededor del 18% de TW esporádicos y sólo una minoría de pacientes con
TW (<5%) tienen mutación germinal de WT1.
Las mutaciones de WT1 se manifiestan a nivel histológico por una tinción
inmunohistoquímica para WT1 anormal o citoplasmática, en comparación con la
tinción normal nuclear (Figura 2).

Page 8 of 30
Figura 2
B.1.2 WT2
El síndrome de Beckwith-Wiedemann (BWS) es un trastorno de
hipercrecimiento que se manifiesta con peso elevado al nacimiento,
macroglosia, organomegalia, hemihipertrofia, hipoglicemia neonatal, defectos
de la pared abdominal, anomalías de la oreja, y predisposición al desarrollo de
TW (5%) y otras neoplasias. La anomalía genética del BWS se localiza en el
cromosoma 11p15, un locus que se denomina WT2, porque la pérdida de
heterocigosidad en este locus se ha detectado en TW. En este locus se localiza
un cluster de genes que tienen un mecanismo de regulación transcripcional
común y se encuentran de manera normal imprintados. Entre los genes del
cluster se encuentran el insulin-like growth factor 2 (IGF2), H19, y p57Kip2. Los
estudios citogenéticos de pacientes con BWS demuestran tanto la duplicación
del locus 11p15 derivado del padre (trisomía paterna) como la presencia de 2

Page 9 of 30
copias del cromosoma 11 paterno sin secuencias del cromosoma materno
(isodisomía uniparental). Estos hallazgos demuestran que el silenciamiento de
los alelos maternos y la concomitante expresión doble de los alelos paternos es
determinante para la expresividad del síndrome. IGF2, es un factor de
crecimiento que normalmente se expresa únicamente4 del alelo paterno, y
cuando se sobrexpresa en animales causa hipercrecimiento.
Otro de los genes del locus, H19 está adyacente a Igf2 pero se expresa
de manera opuesta, esto es, sólo se expresa el alelo materno, y actúa como gen
supresor de tumores en modelos animales. Tanto IGF2 como H19 tienen
mecanismos de regulación comunes y los tumores con pérdida del imprinting
(LOI) de IGF2 a menudo tienen hipermetilación de H19 lo que conlleva su
silenciamiento.
Aparte de la relación entre el locus 11p15 y BWS en cuanto al riesgo de
TW, hay que señalar que en algunos tumores de Wilms esporádicos se
encuentran pérdidas de la región 11p15 sin alteraciones en la región
centromérica de WT1 en el locus 11p13. Curiosamente el LOH en 11p15 en
estos casos también muestra un significante sesgo hacia la pérdida de alelos
maternos, una vez más sugiriendo que el fenómeno de imprinting en esta
región está implicado en la tumorigenésis del TW.
B.1.3 FWT1 y FWT2
La predisposición familiar para padecer TW es rara y sólo afecta a un 1-5%
de los pacientes con TW. El análisis citogenético de los pedigrís de familias
afectas ha desvelado 2 nuevos locus: FWT1 (Familial Wilms’tumour 1) en
17q12–q21 y FWT2 en 19q13.4.

Page 10 of 30
B.1.4 Otros genes
En TW se han descrito otras alteraciones genéticas secundarias como el
LOH en 1p (en 10% de casos) y en 16q (20%), así como ganancias en 1q. Las
pérdidas en 1p y 16q se asocian a mal pronóstico. Otras alteraciones que
también comportan mal pronóstico son la expresión de la telomerasa y TRKB, y
mutaciones en la proteína TP53 que se encuentra en un 5% de tumores de
Wilms. Las mutationes en TP53 se han identificado en el 75% de los TW que
presentan anaplasia (ver más adelante) y específicamente en las áreas donde
los signos histológicos de anaplasia son evidentes. Parece claro pues que el
desarrollo de anaplasia, que está claramente relacionada con mal pronóstico,
depende en gran parte de la adquisición de mutaciones en TP53.
C. Anatomía patológica
La mayoría de los TW son lesiones únicas pero un 6% son bilaterales
(afectan los 2 riñones) y un 12% multifocales en un mismo riñón. El TW clásico
está compuesto por una proporción variable de tipos celulares (trifásico): el
blastema, el estroma, y el epitelial, que como hemos descrito, recapitulan los
diferentes estadios del desarrollo renal (Figura 3).

Page 11 of 30
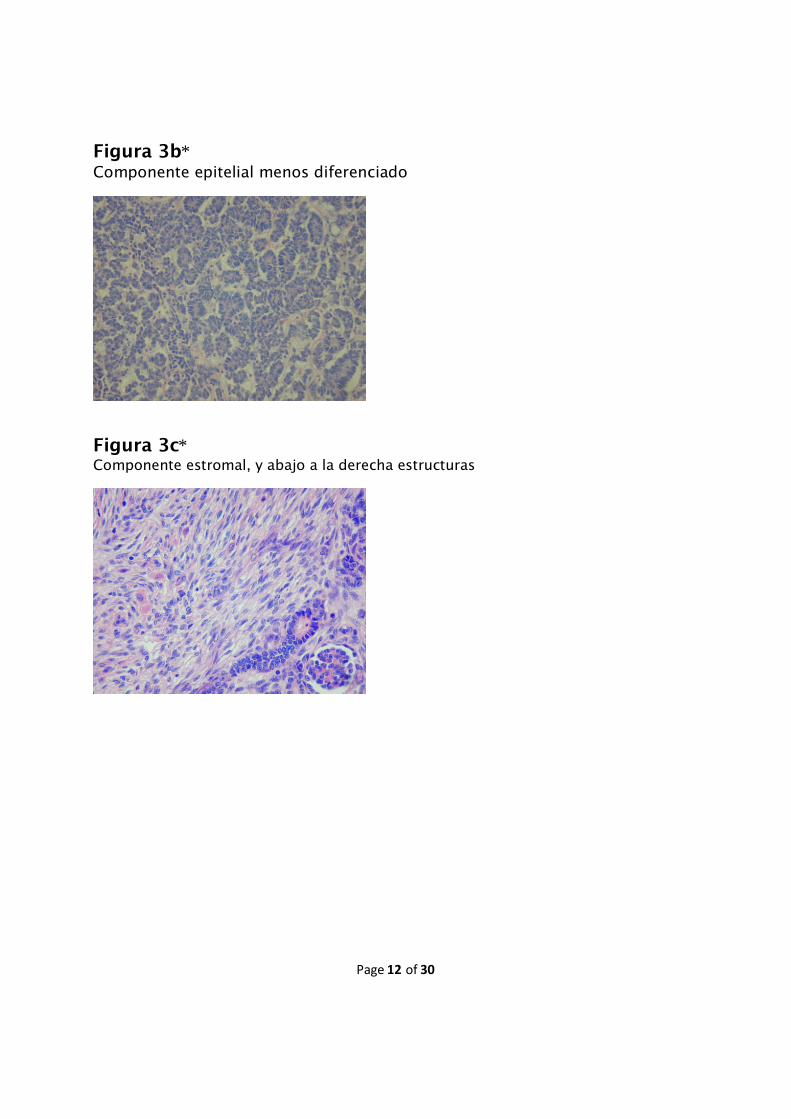
Figura 3*Nefroblastoma Tipo Mixto: Se observan áreas de blastema puro, blastemadiferenciando a epitelio, estructuras epiteliales más maduras de tipo glomeruloides, yademás algo de componenete estromal.
Figura 3a*Estructuras glomeruloides a mayor aumento (componente epitelial mas diferenciado)
Page 12 of 30
Figura 3b*Componente epitelial menos diferenciado
Figura 3c*Componente estromal, y abajo a la derecha estructuras

Page 13 of 30
Figura 3d*Blastema, nidos de diferentes tamaños rodeados de histiocitos.
*Imágenes cerca Dra Laura Galluzzo, Hospital JP Garrahan, Buenos Aires, Argentina
Sin embargo, no todos los TW son trifásicos; algunos son bifásicos y otros
tienen un patrón monofásico. Los TW monofásicos blastematosos suelen ser
muy invasivos y desde el punto de vista histológico presentan dificultades para
distinguirlos de otros tumores embrionarios de la categoría ”small round blue-
cell tumours”, como son el tumor neuroectodérmico primitivo (PNET), el
neuroblastoma, el sarcoma de
Ewing y el linfoma. Igualmente, los TW monofásicos indiferenciados se
pueden confundir con sarcomas como el sarcoma de células claras del riñón, el
nefroma mesoblástico congénito o el sarcoma sinovial. Los patrones
monofásicos epiteliales (raros) pueden ser puramente tubulares o papilares y
pueden ser difíciles de distinguir del carcinoma papilar renal.
Los TW con mutación de WT1 suelen contener en su histología
componentes epiteliales o estromales heterólogos como mucina o epitelio
escamoso, músculo esquelético, cartílago, tejido osteoide, o grasa. Algunos
casos muestran un patrón característico de diferenciación masiva hacia
músculo esquelético y se les conoce como TW rabdomiomatoso o
rabdomioblástico.

Page 14 of 30
La distinción entre TW con histología favorable o desfavorable se basa en la
presencia de signos histológicos nucleares de anaplasia. Ya hemos descrito que
estos cambios morfológicos del núcleo de las células tumorales se
correlacionan con la adquisición de mutaciones en TP53.
Los criterios para el diagnóstico de anaplasia incluyen:
a) figuras mitóticas multipolares o poliploides;
b) tamaños nucleares aumentados al menos 3 veces
los de las células circundantes; y
c) hipercromasia como reflejo del aumento del
contenido de cromatina.
La presencia de anaplasia puede ser focal cuando puede delimitarse bien
la zona del tumor que contiene los signos de anaplasia y no se encuentra en
ninguna región afecta fuera del parénquima renal, o difusa cuando más de
dos zonas están afectas, se encuentra fuera del riñón o bien cuando una sola
biopsia tomada al azar presenta signos de anaplasia. La presencia de anaplasia
se correlaciona con la resistencia a la quimioterapia y no con la agresividad del
tumor. Así, es muy rara en los TW de niños menores de 2 años (estos son
tumores que aparecen en los casos asociados a mutaciones de WT1 y con
histología predominantemente estromal) y va aumentando hasta un 13% en los
TW de niños mayores de 5 años.
Curiosamente, la anaplasia es significativamente más frecuente en
pacientes de raza negra que en los caucásicos. Otros tumores del riñón que
pueden darse en la edad pediátrica, más infrecuentemente, son: el sarcoma de
células claras (Clear cell sarcoma of the kidney o CCSK) que es el tumor más
frecuente después del TW, con un especial tropismo para metastatizar en el
hueso y con un pronóstico más desfavorable; el carcinoma renal (una tercera
parte con translocaciones genéticas características que implican el cromosoma

Page 15 of 30
Xp11.2 y dan lugar a fusiones del gen TFE3); el nefroma mesoblástico
congénito, que es el tumor renal más frecuente en el recién nacido, y que
presenta 2 formas histológicas, la variante clásica y la celular, ésta última con
una translocación específica, t(12;15)(p13;q25) causando la fusión del gen
ETV6 en el cromosoma 12 con el gen NTRK3 en el cromosoma 15; y el tumor
maligno rabdoide renal caracterizado por la inactivación del gen supresor de
tumores HSNF5/INI-1 en el cromosoma 22q dando lugar a una total ausencia en
la tinción inmuno histoquímica para la proteína INI-1 que es diagnóstica para
esta entidad.
C.1 Clinicas
La forma de presentación más común del TW es una masa abdominal
(85%) que por otra parte no causa ningún otro síntoma (Figura 4).
Figura 4*
*Caballero, Emilse. Is Chemotherapy Necessary in a Small, Favorable Wilms Tumor?Oncopedia #100.Released on Oncopedia: 02/08/2008.URL: https://www.cure4kids.org/ums/oncopedia/case_detail/?id=100

Page 16 of 30
Un tercio de los pacientes con la masa abdominal presentan además, dolor,
anorexia, vómitos, malestar general o alguna combinación de éstos. En general,
contrasta la discrepancia entre un tumor grande en el abdomen con el estado
general, relativamente bueno, de muchos de estos pacientes con ausencia de
dolor. En alguna ocasión puede aparecer fiebre que suele ser irregular y pocas
veces elevada. Pueden existir molestias abdominales en forma de dolor sordo,
variable, en posible relación con la micción, por compresión de la masa tumoral
sobre la vía urinaria. A la exploración física se puede detectar hipertensión
arterial en el 25% de los pacientes y anomalías congénitas como aniridia,
malformaciones genitourinarias, hemihipertrofia, o signos de sobrecrecimiento
en el 13–28% de los niños con TW, dependiendo mucho si tienen enfermedad
uni o bilateral. Un 30% de los pacientes presentan hematuria microscópica y
menos del 10% tienen coagulopatía. Se han descrito casos de hipercalcemia
tumoral y poliglobulia por estimulación renal de la síntesis de eritropoyetina.
Cuando el tumor invade la vena renal izquierda y la vena espermática
puede aparecer un varicocele izquierdo. La palpación del abdomen demuestra
la localización de la tumoración en la fosa lumbar con tendencia a crecer hacia
atrás, ocupando la fosa renal, con cierta movilidad que da lugar al “contacto
lumbar”, que sirve para diferenciarlo de la esplenomegalia; también crece hacia
arriba y abajo, adoptando una forma redondeada y ovalada.
La tumoración rara vez sobrepasa la línea media abdominal. La superficie
tumoral es dura, lisa y de bordes redondeados. Ante la sospecha de un TW
debe siempre tenerse en cuenta la precaución de realizar la palpación con
suavidad, para evitar su rotura, y los exámenes complementarios necesarios
con la mayor rapidez. A veces la enfermedad se manifiesta en forma de
abdomen agudo por hemorragia intratumoral, que puede ser espontánea, o
provocada por un traumatismo.
Page 17 of 30
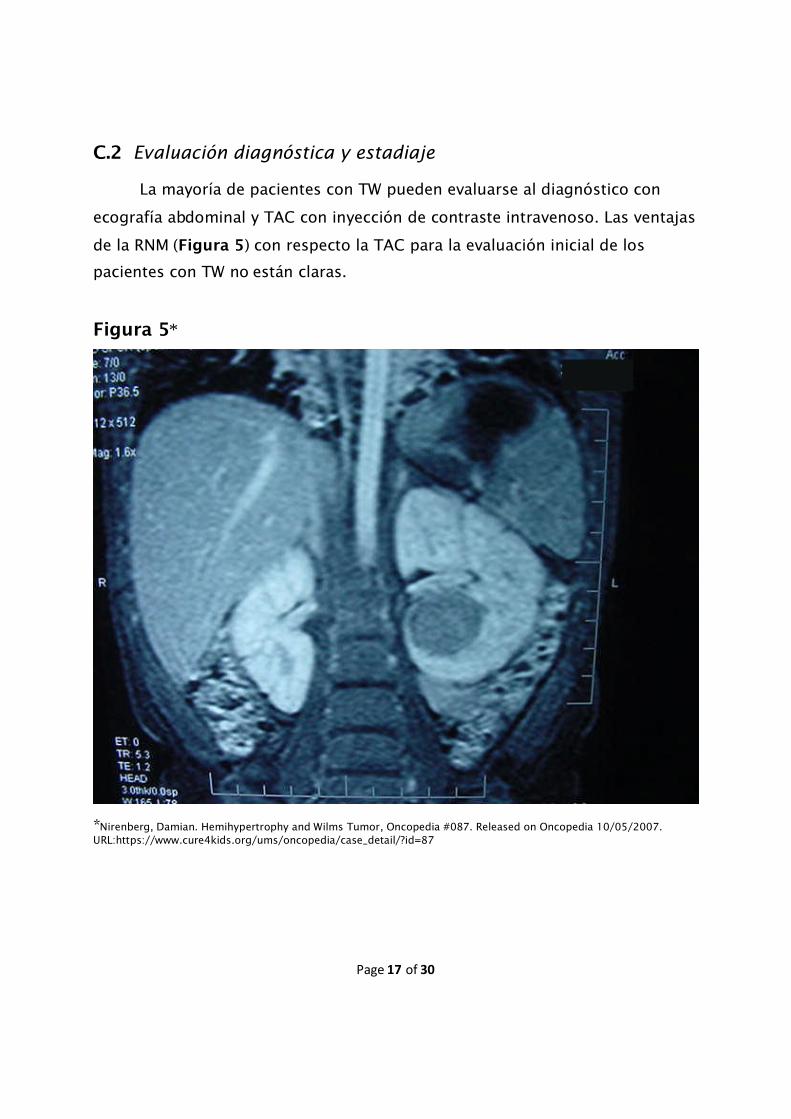
C.2 Evaluación diagnóstica y estadiaje
La mayoría de pacientes con TW pueden evaluarse al diagnóstico con
ecografía abdominal y TAC con inyección de contraste intravenoso. Las ventajas
de la RNM (Figura 5) con respecto la TAC para la evaluación inicial de los
pacientes con TW no están claras.
Figura 5*
*Nirenberg, Damian. Hemihypertrophy and Wilms Tumor, Oncopedia #087. Released on Oncopedia 10/05/2007.URL:https://www.cure4kids.org/ums/oncopedia/case_detail/?id=87

Page 18 of 30
El TW consiste en una masa sólida intrarenal con una pseudo cápsula que causa
una distorsión del parénquima renal y el sistema colector de tipo intrínseco. El
tumor típicamente crece por extensión directa desplazando las estructuras
adyacentes y característicamente no engloba o eleva la aorta abdominal, a
diferencia del neuroblastoma. Puede haber invasión de la vena renal o cava
inferior con extensión a veces hasta la aurícula derecha. Raras veces el tumor
llega a comprimir el uréter lo que causa entonces una gran dilatación piélica,
con hidronefrosis.
El patrón de metástasis es característico y cuando están presentes
aparecen en los pulmones (85%), hígado, y ganglios linfáticos regionales.
En la ecografía, la masa suele ser heterogénea por la presencia habitual de
sangre, grasa, necrosis, o calcificación. El examen de la vena cava inferior es
imperioso para evaluar la extensión del tumor. En la TAC se demuestra la
heterogeneidad de la masa y los ganglios linfáticos regionales así como las
áreas de calcificación y grasa. La inyección del contrate intravenoso sirve para
detector las metástasis hepáticas y ganglionares, la extensión del tumor en la
vena renal o cava inferior, la presencia de un tumor sincrónico contra lateral, y
la presencia de restos nefrogénicos asociados. La imagen del TW por RMN se
caracteriza por una baja señal en T1 y alta señal en T2. La RNM permite
evaluar la presencia de enfermedad multifocal y la permeabilidad de la vena
cava (Figura 5). La evaluación de los restos nefrogénicos o los casos de
nefroblastomatosis generalizada son complejos. La TAC y RNM son claramente
superiores a la ecografía en estos casos.
En la TAC los restos nefrogénicos aparecen como nódulos con baja
atenuación periférica con respecto al parénquima normal. Por RNM los nódulos
presentan una baja señal tanto en T1 como en T2 (a diferencia del TW).
La TAC permite estudiar también la presencia de metástasis hepáticas o
pulmonares. Para descartar afectación ósea en pacientes con metástasis
hematógenas evidentes o con formas histológicas desfavorables, se debe

Page 19 of 30
realizar una gammagrafía ósea. Una diferencia fundamental con el
neuroblastoma es que las metástasis del TW en médula ósea son excepcionales.
Los análisis de sangre en los pacientes con TW suelen mostrar un hemograma
normal, salvo en los casos asociados a hemorragia intratumoral, y una función
renal generalmente sin alteraciones. Por otra parte, no existen marcadores
tumorales para el diagnóstico o seguimiento de posibles recidivas. En el análisis
de orina es posible encontrar, incluso sin hematuria macroscópica, algunos
hematíes, leucocitos y piocitos; en muy contadas ocasiones, han sido
demostradas células tumorales.
D. Estadiaje
El TW se estadia en función de la extensión anatómica sin
consideraciones de tipo biológico o genético. Actualmente se utilizan dos
sistemas de estadiaje (y manejo) del TW: uno basado en la cirugía inicial antes
de ningún tratamiento con quimioterapia, desarrollado por el National Wilms’
Tumor Study Group (NWTSG) en USA, y el otro basado en la respuesta a la
quimioterapia previa a la cirugía desarrollado por el International Society of
Pediatric Oncology (SIOP) en Europa. Ambos sistemas de estadiaje han
demostrado su capacidad para estratificar la terapia y clasificar a los pacientes
en función del pronóstico. Hay que resaltar, sin embargo, que las diferencias
en el tiempo quirúrgico en ambos sistemas confunde las comparaciones estadio
por estadio en ambos sistemas.
D.1 Estadio 1
NWTSG: El tumor se encuentra confinado en el riñón y se reseca
completamente sin afectación de la cápsula renal o de los vasos del seno renal.
El tumor no se rompe antes de su extracción.

Page 20 of 30
SIOP: El tumor se encuentra confinado en el riñón. La cápsula puede estar
afectada pero su superficie externa está libre de tumor y se reseca
completamente. El tumor puede protruir en la pelvis y el uréter pero no los
infiltra. Los vasos del seno renal no están invadidos.
D.2 Estadio II
NWTSG: El tumor se extiende más allá del riñón pero se puede resecar
completamente. Hay extensión regional del tumor bien en el seno renal o la
cápsula. Los vasos del parénquima renal o seno renal pueden estar invadidos. El
tumor se desparrama durante la biopsia o la extracción pero la diseminación se
confina al flanco, sin afectar el peritoneo. No hay evidencia de enfermedad más
allá de los márgenes de resección (los ganglios linfáticos regionales biopsiados
deben ser microscópicamente negativos). Se efectúa biopsia del tumor antes de
la resección quirúrgica.
SIOP: El tumor se extiende más allá del riñón pero se puede resecar
completamente (márgenes de resección “libres”).
Los vasos del parénquima renal o seno renal pueden estar invadidos así como
los ganglios linfáticos y los órganos adyacentes como la vena cava, pero son
resecados completamente.
D.3 Estadio III
NWTSG: Después de la cirugía queda tumor en el abdomen y su origen no
es por diseminación hematógena:
A. Ganglios linfáticos patológicos en abdomen o pelvis;
B. El tumor penetraba a través de la superficie peritoneal; C. Implantes
tumorales en la superficie peritoneal;
D. Queda tumor micro o macroscópico en el abdomen después de
la cirugía;

Page 21 of 30
E. El tumor se considera inoperable por infiltración de órganos vitales;
F. El tumor se desparrama más allá del flanco.
SIOP: Después de la cirugía queda tumor en el abdomen y su origen no es
por diseminación hematógena:
A. Resección incompleta que se extiende micro o macroscópicamente más allá
de los márgenes de resección;
B. Ganglios linfáticos patológicos en abdomen o pelvis;
C. Ruptura del tumor antes o durante el acto operatorio; D. El tumor penetraba
a través de la superficie peritoneal;
E. Trombo tumoral presente en los márgenes de resección de los vasos o
uréter, trans-seccionado o resecado en fragmentos;
F. El tumor se ha biopsiado quirúrgicamente antes de la quimioterapia
preoperatoria.
D.4 Estadio IV
NWTSG y SIOP: Presencia de metástasis hematógenas (pulmón, hígado, hueso,
cerebro) o ganglios linfáticos fuera de la cavidad abdomino-pélvica.
D.5 Estadio V
NWTSG y SIOP: TW bilateral.

Page 22 of 30
E. Tratamiento
El manejo del TW históricamente ha sido diferente en Europa (el grupo
SIOP) y en los EE.UU. (el grupo NWTS). Excepto en los pequeños tumores en los
que puede iniciarse el tratamiento con la intervención quirúrgica, el grupo
Europeo ha recomendado en todos los demás estadios una quimioterapia
reductora, sin biopsia, antes de la intervención quirúrgica. Una vez practicada la
extirpación, se valora el estadio nuevamente y se determina el riesgo asociado
al tipo histológico (grado de respuesta a la quimioterapia preoperatoria).
Por su parte, el grupo americano ha promovido la práctica de la intervención
quirúrgica o la biopsia al diagnóstico pues considera imperativo la obtención
del diagnóstico histológico y el estadiaje.
La estrategia SIOP se ha fundamentado históricamente en 1) evitar el
riesgo quirúrgico de la nefrectomía inmediata al diagnóstico pues los tumores a
menudo son grandes en su presentación; y 2) disminuir las posibilidades de
diseminar el tumor en la zona del flanco por la biopsia. La estrategia SIOP es
controvertida pues pacientes con imágenes típicas y características clínicas
sugestivas de TW se tratan con quimioterapia sin confirmación histológica. Los
resultados de las series SIOP demuestran que un 1% de casos tratados tienen
patologías no cancerosas y un 12% tienen otras neoplasias. Por otra parte, los
estudios más recientes demuestran que la biopsia percutánea con aguja
fina no afecta el estadiaje del tumor ni el tratamiento.
La ventaja más importante de la estrategia SIOP ha sido que la
quimioterapia preoperatoria reduce efectivamente el volumen del tumor en la
mayoría de los casos, diminuyendo las posibilidades de rotura del tumor y
diseminación y además, bajando el estadiaje postoperatorio. Así, en los
estudios SIOP más recientes menos pacientes recibieron radioterapia local en el
flanco comparado con el último estudio del NWTS-5.
La segunda ventaja de la estrategia SIOP ha sido comprobar que la
respuesta a la quimioterapia preoperatoria constituye un factor pronóstico

Page 23 of 30
significativo. Otra diferencia notable entre los grupos NWTSG y SIOP ha sido el
manejo de las metástasis pulmonares. En el último estudio del NWTSG los casos
con imágenes nodulares compatibles con metástasis, pero visibles sólo en la
TAC, se trataron con radioterapia pulmonar. La supervivencia de estos
pacientes en el NWTS-4 fue del 81%.
En los estudios SIOP, sólo se valoraban como metástasis pulmonares las
lesiones pulmonares visibles en la radiografía simple de tórax. Si las lesiones
pulmonares desaparecían completamente con la quimioterapia, o se resecaban
quirúrgicamente, los pacientes no recibían radioterapia pulmonar. Así, la
supervivencia a los 4 años reportada para estos pacientes en el estudio SIOP
más reciente fue del 83%. Estos resultados ponen en cuestión la necesidad de
radioterapia pulmonar en los casos con buena respuesta a la quimioterapia.
A pesar de las diferencias históricas en el manejo del TW, no existen
diferencias significativas en los resultados entre ambos grupos y hoy en día
también el grupo SIOP promueve en todos los casos la biopsia al diagnóstico.
F.1 Cirugia
La exéresis del tumor deberá ser practicada en todos los casos. El
tratamiento quirúrgico de elección es la nefrectomía completa (Figura 4). Las
nefrectomías parciales están indicadas en menos del 5% de los casos, incluso
después de la reducción del tamaño tumoral con quimioterapia preoperatoria,
pues la mayoría de los TW son muy grandes o localizados centralmente.
Técnicamente se recomienda un abordaje transperitoneal para permitir una
exposición adecuada para realizar el estadiaje quirúrgico local. Ello incluye
la mobilización e inspección del riñón contralateral para excluir enfermedad
bilateral, antes de realizar la nefrectomía. Permite, además, la inspección de los
ganglios linfáticos regionales y del hilio renal que deben siempre biopsiarse
cuando aparezcan sospechosos pues constituyen un elemento clave para el

Page 24 of 30
estadiaje. La resección del tumor infiltrando parcialmente órganos como
pueden ser el diafragma, hígado, o el músculo psoas debe realizarse si se
consigue una resección completa con poca morbilidad. Ello permite convertir
un estadio III en estadio II, si los márgenes están libres, con la consiguiente
reducción de la terapia citotóxica. La extensión del tumor en la vena renal o
cava inferior próxima al riñón puede resecarse habitualmente en bloque
con el riñón. La resección de la cava inferior hasta el hilio hepático o la aurícula
derecha incrementa notablemente la morbilidad quirúrgica y no debería hacerse
en el mismo procedimiento inicial. En estos casos la quimioterapia
preoperatoria disminuye el tamaño y extensión del tumor y el trombo tumoral,
sin que aumente su adherencia a la pared vascular, facilitando la resección
quirúrgica.
F.2 Tratamiento Citotóxico (Quimioterapia y radioterapia)
Tres fármacos han demostrado una efectividad indisputada en el
tratamiento del TW desde los años 1950s y 1960s la actinomicina-D y la
vincristina, que son los pilares del tratamiento, y la doxorubicina que se añadió
en los años 1970s. A lo largo de las décadas la combinación de estos tres
fármacos, la duración del tratamiento y la manera de administrarlas se han
refinado para maximizar la supervivencia de los pacientes a la vez que se han
reducido los efectos secundarios agudos y a largo término. Otros cuatro
quimioterápicos se utilizan en pacientes de alto riesgo, que recaen o no
responden inicialmente a la combinación de actinomicina-D, vincristina, y
doxorubicina, son la ciclofosfamida, la ifosfamida, el carboplatino, y el
etopósido.
En el caso de aplicar el protocolo SIOP, la quimioterapia prequirúrgica
consiste en actinomicina D y vincristina semanales, durante 4 semanas.
Después de la cirugía, el tratamiento citotóxico oscila desde finalizar el
tratamiento en caso de estadio Ipostquirúrgico con bajo riesgo, a pautas de

Page 25 of 30
tiempo variables (de 4 a 34 semanas) según los estadios y riesgos histológicos,
con vincristina y actinomicina D asociadas o no a adriamicina, etopósido,
ciclofosfamida y carboplatino. Cuando persisten restos tumorales o la
histología es desfavorable, se añade radioterapia en el lecho tumoral.
La radioterapia continua siendo importante en el tratamiento del TW. Los
protocolos estándar de radioterapia incluyen la irradiación del flanco abdominal
con 10,8 Gy en seis fracciones para los estadios III con histología favorable y
estadios II–III con anaplasia difusa. La radioterapia pulmonar se administra si
tras el primer ciclo de quimioterapia postquirúrgica siguen visibles las
metástasis. Los enfermos con estadio IV (metástasis hematógenas) reciben
pautas de quimioterapia más prolongadas y agresivas. En algún caso de
metástasis pulmonar localizada, en uno o ambos pulmones, es posible la
exéresis quirúrgica, conservando al máximo el tejido pulmonar sano.
La enfermedad refractaria y las recidivas se tratan combinando cirugía,
radioterapia y quimioterapia.
Combinaciones de quimioterápicos como ICE (ifosfamida, carboplatino,
y etopósido), han conseguido mejorar la supervivencia después de recaídas de
manera significativa hasta niveles del 50%–60%. En los pacientes que responden
a la quimioterapia de rescate, es posible emplear tratamientos de consolidación
con autotrasplante de células progenitoras hematopoyéticas.
Alrededor del 6% de los pacientes con TW se presentan al diagnóstico con
tumores simultáneos (sincrónicos) en los dos riñones. Aunque más del 70%
sobreviven, estos niños tienen un gran riesgo para desarrollar fallo renal
terminal. Este riesgo ha hecho que las pautas de manejo del estadio V de Wilms
recomienden un estadiaje bilateral, cada riñón por separado, sin resección
quirúrgica inicial, seguido de quimioterapia para reducir el tamaño de los
tumores y así facilitar procedimientos quirúrgicos que puedan ser más
conservadores dejando parénquima renal sano. El tratamiento se hace en
función del estadio local de cada riñón y de la histología. Después de 6–8
semanas de quimioterapia se revalúa el paciente y la posibilidad de resección

Page 26 of 30
quirúrgica conservadora. En algunos casos un procedimiento de second-look
puede estar indicado para evaluar la posibilidad de resecar el tumor
preservando tejido renal. Más quimioterapia o radioterapia pueden ser
necesarias pero en general la cirugía definitiva debería realizarse no más allá de
las semanas 12–16 del diagnóstico para evitar la aparición de resistencias.
Las supervivencias alcanzadas por los grandes grupos cooperativos para
los pacientes con cualquier estadio e histología favorable son iguales o
superiores al 80% (95% para el estadio I y 80% para el estadio IV). Para los
pacientes con anaplasia, sin embargo, los estudios más recientes, como el
NWTS-5, muestran una supervivencia a los 4 años para los pacientes con
anaplasia difusa del 55% y para la anaplasia focal, del 75%. Los pacientes con
estadio II, III, y IV con anaplasia y nefrectomía inmediata tuvieron una
supervivencia del 82%, 68%, y 37%, respectivamente.
F.3 Secuelas
A medida que mayor porcentaje de niños con TW se curan, aparecen
nuevos datos en relación a las secuelas directa o indirectamente relacionadas
con la terapia. El tipo de secuelas depende de la edad y el sexo, el tipo de
cirugía practicada, los quimioterápicos utilizados, y la radioterapia. Los órganos
de mayor riesgo incluyen los riñones, el corazón, y las gónadas.
La causa más frecuente de fallo renal es la nefrectomía bilateral, y la segunda
el daño inducido por la radioterapia y las complicaciones quirúrgicas. La
frecuencia de fallo renal en los casos de TW bilateral ha evolucionado desde el
16% en la década de los 1980s hasta el 3-8% en el estudio NWTS-4 en los
1990s. El fallo cardiaco congestivo es una complicación conocida de la
administración de antraciclínicos (adriamicina) y el riesgo se incrementa con la
administración concomitante de radioterapia pulmonar. Consecuentemente, los
pacientes con TW que reciben adriamicina deben ser monitorizados a largo
término. La función pulmonar puede afectarse asimismo en aquellos pacientes
que reciben radioterapia pulmonar, sobretodo cuando se trata de forma

Page 27 of 30
bilateral. En estos pacientes, la capacidad pulmonar total puede reducirse hasta
en un 50–70%. La función gonadal puede afectase en las niñas que reciben
radioterapia abdominal por TW. Los estudios del NWTSG han demostrado que
estas mujeres están en mayor riesgo de padecer abortos o embarazos
complicados.
Finalmente algunos de los niños tratados por TW están en especial riesgo
para desarrollar segundas neoplasias, ya sea por el efecto carcinogénico del
tratamiento recibido o bien como resultado de una predisposición hereditaria a
desarrollar tumores. La incidencia acumulada de segundas neoplasias en los
estudios más recientes del NWTSG son del 1,6% a los 15 años. La mayoría de
estos tumores aparecen en zonas previamente irradiadas. El mayor factor de
riesgo es la radioterapia, aunque los pacientes que sólo recibieron vincristina y
actinomicina también tienen un riesgo más elevado cuando se les compara con
la población sana.
G. Otros tumores renales
Sarcoma de células claras del riñón. (Clear cell sarcoma of the kidney o
CCSK) es el tumor renal más frecuente después del TW, y representa un 5% de
las neoplasias renales infantiles. Tiene un especial tropismo para metastatizar
en el hueso (50% de los metastásicos) pero también en sistema nervioso
central. Su pico de incidencia es entre 1–4 años, predominantemente afectando
varones, y su forma de presentación más común es la masa abdominal. No se
han reportado casos de afectación bilateral. El pronóstico es más desfavorable
que el TW y el tratamiento habitualmente consiste en los regímenes diseñados
para el TW de alto riesgo.
El carcinoma renal supone un 2–5% de todos los tumores renales
pediátricos, siendo la neoplasia renal más frecuente en el adulto. La media de
edad al debut es entre los 9 y 15 años, claramente mayores que el TW. Los

Page 28 of 30
pacientes suelen presentar con hematuria, dolor, y una masa palpable, aunque
algunos pueden ser asintomáticos, una presentación similar al adulto. Las
metástasis al diagnóstico se detectan en un 20% de los casos, habitualmente en
los pulmones, hueso, hígado, o cerebro. Las presentaciones bilaterales y las
presentaciones en niños pueden asociarse a síndromes predisponentes como la
enfermedad de von Hippel-Lindau, el carcinoma renal papilar hereditario, el
oncocitoma familiar asociado con el síndrome Birt-Hogg-Dubey y el carcinoma
renal hereditario.En el carcinoma renal, una tercera parte de los tumores tienen
translocaciones genéticas características que implican el cromosoma Xp11.2 y
dan lugar a fusiones del gen TFE3.
La supervivencia de los pacientes con carcinoma renal depende en gran
medida de la posibilidad de realizar o no una nefrectomía radical, con una
supervivencia global del 64–87%. A los 5 años, la supervivencia para el estadio I
es del 90%, 50–80% para estadios II–III, y menos del 10% para la enfermedad
metastásica o estadio IV.
El nefroma mesoblástico congénito (tumor de Bolande), es el tumor renal
más frecuente en el recién nacido, y tiene un comportamiento, en general,
benigno. Existen 2 formas histológicas, la variante clásica y la forma celular,
ésta última con una translocación específica, la t(12;15)(p13;q25) que causa la
fusión del gen ETV6 en el cromosoma 12 con el gen NTRK3 en el cromosoma
15. El tratamiento es principalmente quirúrgico y debe ser radical para evitar
posibles recidivas.
El tumor teratoide-rabdoide maligno del riñón, representa un 2% de los
tumores renales del niño, y es el más agresivo de todos ellos. Afecta
predominantemente a niños menores de un año. El tumor maligno rabdoide
está caracterizado por la inactivación del gen supresor de tumores HSNF5/INI-1
en el cromosoma 22q dando lugar a una total ausencia de expresión en la
tinción inmunohistoquímica para la proteína INI-1 que es diagnóstica para esta
entidad.

Page 29 of 30
Debe descartarse en estos casos una alteración constitucional del gen
HSNF5/INI-1, que predispone a la aparición de la neoplasia, tanto en el riñón
como en otros órganos, principalmente el sistema nervioso central. El
tratamiento debe ser radical, combinando cirugía, radioterapia y quimioterapia,
a pesar de lo cual la supervivencia no supera el 25% a los 4 años.
Incluso los casos con resección quirúrgica completa y ganglios linfáticos
negativos tienen una supervivencia sólo del 50% con los tratamientos derivados
de protocolos de TW.
Recientemente se han diseñado protocolos específicos para tumores
rabdoides con más intensidad de agentes alquilantes y antraciclinas, y parece
que consiguen mejores respuestas iniciales.
Los tumores benignos, aunque raros, pueden presentarse en el riñón en
desarrollo. Debe recordarse, entre otros, el quiste hidatídico, el hamartoma
fibromatoso, el angiolipoma, el hemangioma, el adenoma, el teratoma y las
malformaciones quísticas renales.
Referencias Bibliográficas
Ahmed HU, Arya M, Levitt G, Duff PG, Mushtaq I, Sebire NJ. Part I: Primarymalignant non-Wilms’ renal tumours in children Lancet Oncol 2007; 8: 730–37.
Ahmed HU, Arya M, Levitt G, Duff y PG, Sebire NJ, Mushtaq I. Part II: Treatment ofprimary malignant non-Wilms’ renal tumours in children. Lancet Oncol 2007: 8: 842–48.Golden CB, Feusner JH. Malignant abdominal masses in children: quick guide toevaluation and diagnosis. Pediatr Clin North Am 2002; 49: 1369-92.
Graf N, Tournade MF, de Kraker J. The role of preoperative chemotherapy in themanagement of Wilms’ tumor. The SIOP studies. International Society of PediatricOncology. Urol Clin North Am 2000; 27: 443-54.
Grundy PE, Green DM, Coppes MJ et al. Renal Tumors. En: Pizzo PA, Poplack DG.Pediatric Oncology. Filadelfia: Lippincott Williams and Wilkins, 2002. p. 865-94.
Morales LL, Cruz Martínez O. Tumor de Wilms. En: Cruz M. Tratado de Pediatría. 8ªed. Madrid: Ergon, 2001. p. 1499 (con citas bibliográficas de años anteriores).

Page 30 of 30
Neville HL, Ritchey ML. Wilms’ tumor. Overview of National Wilms’ Tumor StudyGroup results. Urol Clin North Am 2000; 27: 435-42.
Pritchard-Jones K, Pritchard J. Success of clinical trials in childhood Wilms’ tumoraround the world. Lancet 2004; 364: 1468-70.
Rivera MN, Haber DA. Wilms’ tumor: connecting tumorigenesis and organdevelopment in the kidney. Nat Rev Cancer 2005; 5: 699-712.
Royer-Pokora B, Beier M, Henzler M et al. Twenty-four new cases of WT1 germlinemutations and review of the literature: genotype/phenotype correlations for Wilms tumordevelopment. Am J Med Genet A 2004; 127: 249-57.







