

UTILIDAD DE LOS ESTUDIOS GENÉTICOS EN LAS ENFERMEDADES HEPÁTICAS SIN
HERENCIA MENDELIANA
FECHA REALIZADO REVISADO APROBADO NOMBRE Consejo Calidad CARGO
FIRMA
Lugar de archivo : Dirección Responsable custodia: Fecha de revisión :
PLAN DE ACOGIDA
Prof. Manuel Romero Gómez. UGC IC Aparato Digestivo.
Instituto de Biomedicina de Sevilla. Ciberehd. HU Virgen del Rocío. Universidad de Sevilla
CURSO DE POSGRADO“Aspectos diagnósticos y terapéuticos controvertidos en hepatología”
Genética, colestasis e hígado grasoDIRECTOR:
RAÚL J. ANDRADE BELLIDO

Introduction
Karlsen, T et al. J Hepatol 2015; Haley, K. Int J Gen 2015; Eslam J Hep Int 2016
by linkage disequilibrium and haplotype analysis in more than100 families [8]. Functional studies demonstrated that it encodesa P-type ATPase gene with metal binding regions similar to thosefound in prokaryotic heavy metal transporters [9]. Building onthis discovery, subsequent studies were able to determine vastaspects of previously unknown aspects of copper transport andthe pathophysiology of Wilson disease [43]. Using a similarstrategy three years later, the HFE gene in the extended majorhistocompatibility complex (MHC) was shown to be mutated inpatients with autosomal-recessive haemochromatosis [10].Furthermore, additional types of non-HFE hereditary haemochro-matosis were subsequently described, linked to mutations in theferroportin gene (SLC40A1) [17,18], the transferrin receptor 2gene (TFR2) [19], the hepcidin gene (HAMP) [20] and the hemoju-velin gene (HJV) [21], respectively. These genetic discoveriesjointly paved the way for the full characterization of hepatic ironmetabolism and its regulators [44], allowing for dissection ofmechanisms responsible for the development of liver disease inthe presence of detected mutations.
The mutation profile of disease genes varies. Hereditaryhaemochromatosis is most often caused by a predominantfounder mutation (p.C282Y) which accounts for approximately95% of patients [45]. Wilson disease shows a different geneticprofile, with extensive allelic heterogeneity with more than 500mutations described in the ATP7B gene to date. For this reason,genetic testing in haemochromatosis is undertaken via targetedgenotyping of the predominant variants (always starting withp.C282Y), whereas in Wilson disease there is a need for genesequencing and variant analysis to obtain genetic support forthe diagnosis.
The first GWAS in hepatobiliary diseases was performed forgallstone disease and confirmed the candidacy of the hepatobil-iary cholesterol transporter ABCG5/G8 as a major susceptibilitygene for gallstones worldwide, with p.D19H representing thelikely causal single nucleotide polymorphism (SNP) [22,46].Other rare loss-of-function SNVs in this transporter had beenidentified previously, in individual patients, to underlie themonogenic disease sitosterolemia, which is characterized byunrestricted intestinal absorption of both cholesterol andphytosterols such as sitosterol [47]. The adjacent, oppositely ori-ented ABCG5/G8 genes encode two ATP-binding cassette (ABC)hemitransporters that are localized in the apical membranes ofenterocytes and hepatocytes. The case also illustrates a
Table 1. Present day technologies involved in the detection of disease relevant genetic variation. These genomic technologies are principally deriving from a variety ofsequencing techniques (targeting genes, the coding regions of the genome [exome] or the whole genome) and microarrays for genome-wide single nucleotide or copynumber variant detection. Cost (price) and the availability of relevant bioinformatic tools also need considerations in making the ideal selection of method for investigatingany observed trait.
Mendelian diseases Complex diseasesDegree of heritability Relative sibling risk increased >200-1000 times
compared with general population riskRelative sibling risk increased 2-20 times compared with general population risk
Technology preferences Whole-exome and whole-genome sequencing Single nucleotide polymorphism arraysStudy design Linkage analysis, in silico candidate variant filtering and
prioritization, functional assessments of disease variantsGenome-wide case-control association analysis
Key challenges Large number of candidate variants makes conclusive identification difficult, mechanistic support for causality needed and may require extensive studies
Low effect size of relevant variants means large numbers of patients and controls are needed to establish robust findings (typically thousands)
Genetics
Pathophysiology ATP7B, HFE, JAG1, ABCC2, ABCG5/8
ABCB4, OATP1B1/3, TJP2
ATP8B1, ABCB11, ABCB4, PNPLA3
ABCB4, ATP7B, HFE, JAG1
IL28B/IL29
Genes (examples)
Definition
Classification
Diagnosis
Treatment
Fig. 1. Scope of review. With an emphasis on the developments of the last30 years, the present article aims to discuss the implications of genetic studies inliver diseases onto relevant aspects of research and clinical hepatology. Topicscovered range from descriptions of basic pathophysiology to practical aspects ofdiagnosis and treatment of the individual patient. The examples have beenselected to illustrate the utility of genetic tools for specific aspects of clinicalpractice. For details, see text; for gene name abbreviations, see http://www.ncbi.nlm.nih.gov/gene/.
Key Points
Different liver diseases are at different stages of the following sequence of steps:
• The first genetic discoveries identified the major loci for Mendelian diseases
• Numerous studies have identified susceptibility to complex diseases
• All diseases are actually complex with multiple genes contributing to the phenotype
• With the identification of different underlying genetic contributors to disease those diseases are being reclassified
• A better understanding of genetic aetiology will suggest new modes of treatment for disease
• A complete understanding of all the contributors to a individual’s disease will allow personally tailored treatment
JOURNAL OF HEPATOLOGY
Journal of Hepatology 2015 vol. 62 j S6–S14 S7
Virus/NAFLD/ALD/DILI/AIH/PBC/PSC
HH/EW/A1AT

Candidate Gene
Genetic approaches
GWAS (Hypothesis free)
Association accuracy
Sample size
Biological process
Validation in external cohorts
SANGER Seq >> PCR >> NGSKarlsen, T et al. J Hepatol 2015

Spontaneous viral clearance
Fibrosis progression
Response to therapy
Cirrhosis outcomes
years
HCVHBVDrugsDietAlcohol
Hepatocyte chronic infection?Hepatocyte steatosis?Liver damage?
Fibrosis progression to cirrhosis Liver cancer
Cirrhosis outcomes:Variceal bleedingAscitesHE
Response to therapy
Cure
G E N E S
Kupffer cellsCells of the resident hepatic macrophage population. In the steady state, Kupffer cells are thought to self-renew and to originate from fetal (yolk sac) precursor cells. These cells reside in the liver sinusoids where they regulate local immune responses and remove bacteria, bacterial endotoxins and microbial debris that are derived from the gastrointestinal tract and transported to the liver via the portal vein.
FibrocytesCells of haematopoietic origin (marked by CD45 expression) that can differentiate into tissue myofibroblasts.
PericytesSpecialized mesenchymal cells that are embedded within the basement membrane of capillaries (hepatic stellate cells are considered to be the pericytes of the liver).
Space of DisseThe perisinusoidal space in the liver, between the endothelial cells and the hepatocytes.
Hepatic stellate cells. Hepatic stellate cells are the pericytes of the liver and they reside in the space of Disse between the hepatocytes and the endothelial cells, where they encircle the liver sinusoids. They express neural markers (such as glial fibrillary acidic protein (GFAP) and synaptophysin) and desmin, and they store vitamin A in lipid droplets. Following chronic liver injury of any cause, hepatic stellate cells become acti-vated and transdifferentiate to myofibroblast-like cells, which are characterized by a loss of lipid droplets, by increased proliferation and migration, by secretion of excessive ECM proteins, by enhanced contractility and by the release of pro-inflammatory and pro-fibrogenic factors including TGFβ. Activated hepatic stellate cells upregulate mesenchymal cell markers (such as αSMA and type I collagen) and lose their neural marker signa-ture, which enables the cellular activation status of these cells to be discriminated in vitro and in vivo.
Perpetuation of hepatic myofibroblast activationPerpetuation of myofibroblast activation results from several positive feedback loops involving, among other receptors, TGFβ receptors, PDGF receptor-β15 and angiotensin II receptors16, which are upregulated on these cells. In addition, TGFβ promotes myofibroblast survival through the activation of focal adhesion kinase (FAK) and AKT17. The production of soluble mediators
such as CC-chemokine ligand 2 (CCL2; also known as MCP1) and macrophage colony-stimulating factor (M-CSF) augments inflammatory cell infiltration to the site of injury to initiate and to maintain myofibroblast activation. Myofibroblast differentiation and fibrosis may also be promoted by epigenetic events such as the methyl-CpG-binding protein 2 (MeCP2)-mediated silencing of the gene encoding peroxisome proliferator-activated receptor-γ (PPARγ)18 or the transcriptional activation of pro-fibrogenic genes by specific histone modifications19. An inevitable consequence of fibrotic ECM accumulation (and myofibroblast contraction) is a progressive increase in tissue stiffness. There is evi-dence showing that biomechanical signalling, medi-ated through increased tissue stiffness, is a crucial mechanism to promote and to sustain the differentiated, contractile myofibroblast phenotype20 and to stimulate the force-dependent activation of TGFβ by dissociating it from latency-associated peptide (LAP)21.
Cell adhesion proteins such as integrins mediate complex cell–cell and cell–ECM interactions in wound-healing responses. Integrins transduce bidirectional sig-nals that regulate cell behaviour, including proliferation, motility, differentiation, survival and apoptosis. During fibrogenesis, increased expression of αv integrins on hepatic myofibroblasts22 and of αvβ6 integrin on acti-vated cholangiocytes modifies the cellular response to
Figure 1 | Natural history of chronic liver disease. Hepatic fibrosis is the wound-healing response of the liver to many causes of chronic injury, of which viral infection, alcohol and non-alcoholic steatohepatitis (NASH) are the most common. Regardless of the underlying cause, iterative injury causes inflammatory damage, matrix deposition, parenchymal cell death and angiogenesis leading to progressive fibrosis. The scar matrix typically accumulates very slowly (the median time to cirrhosis in chronic hepatitis C is 30 years) but once cirrhosis is established the potential for reversing this process is decreased and complications develop. Genetic polymorphisms, epigenetic marks and cofactors (such as obesity and alcohol) can modulate the risk of fibrosis progression. If the cause of fibrosis is eliminated, resolution (that is, complete reversal to near-normal liver architecture) of early hepatic fibrosis can occur. In cirrhosis, although resolution is not possible, regression (that is, improvement but not reversal) of fibrosis improves clinical outcomes. Anti-fibrotic therapies are emerging that can slow, halt or reverse fibrosis progression. Currently, liver transplantation is the only available treatment for liver failure or for some cases of primary liver cancer. Hepatocellular carcinoma is rising in incidence worldwide and is a major cause of liver-related death in patients with cirrhosis.
Nature Reviews | Immunology
Normal liver
Earlyfibrosis
Resolution Regression
?
Cirrhosis
Livertransplant
Hepatocellularcarcinoma
n ammatory damage atri deposition arenchymal cell death Angiogenesis
enetic polymorphisms pigenetic mar s Cofactors (such as obesity and alcohol)
Chronic injury iral infection lcohol NASH utoimmune disorders Cholestatic disorders etabolic diseases
isrupted architecture oss of function berrant hepatocyte regeneration
iver failure ortal hypertension
0 years
emoval of underlying cause nti- brotic drug or cell therapy
R E V I E W S
NATURE REVIEWS | IMMUNOLOGY VOLUME 14 | MARCH 2014 | 183
F O C U S O N h O m E O S tat I C I m m U N E R E S p O N S E S
© 2014 Macmillan Publishers Limited. All rights reserved

PNPLA3rs738409
ASHNASH
Stickel, F et al. J Hepatol 2017; Liu et al 2014 Nat Commun 2014
11090 October 21, 2015|Volume 21|Issue 39|WJG|www.wjgnet.com
in animal models and human hepatocytes[19]. The I148M variant - a SNP with a risk allele frequency of 21%-28% in European populations - impairs the phospholipase activity of the enzyme, thus reducing lipid catabolism, although it might also gain new functions[17] with a resulting increase in the synthesis of phosphatidic acid[20]. In addition, the PNPLA3 variant has been associated with a loss of retinyl-palmitate lipase activity in stellate cells[21]. Taken together, these data support a link between the PNPLA3 variant and the above reported wide spectrum of liver damage. As previously mentioned, the first report on the PNPLA3 I148M variant in NAFLD came from the GWAS by Romeo et al[15]. These authors identified the relationship between this SNP and liver fat content, and this association remained significant after adjusting for metabolic factors, ethanol use, and ancestry. Of great relevance, the link between PNPLA3 I148M variant and NAFLD is not confounded by the presence of metabolic syndrome (MS) and its features; indeed, even if some authors reported an interplay
between insulin resistance (IR) and the variant[22,23], most studies did not find such association, as confirmed by a recent meta-analysis[24]. Interestingly, this independent association between the PNPLA3 I148M variant and NAFLD could be more relevant in women than in men, as highlighted by Speliotes et al[25] in a gender specific analysis performed on a histological NASH cohort. Beyond these gender differences, however, the PNPLA3 I148M variant could explain, at least in part, the variations in NAFLD prevalence across different multiple ethnicities. Indeed, the original report by Romeo et al[15] already found that the frequencies of the 148M allele matched the prevalence of NAFLD in the Dallas Heart Study[11], such that Hispanics had the highest frequency of the 148M allele (49%), followed by European Americans (23%) and African Americans (17%). These ethnic differences were subsequently confirmed by other investigators[26]. Over the last few years, several studies not only have further emphasized how the PNPLA3 I148M variant is associated robustly with liver fat content[27,28]
Figure 1 Hematic overview of the main genetic variants potentially involved in nonalcoholic fatty liver disease/nonalcoholic steatohepatitis susceptibility and progression. GWAS: Genome-wide association studies; HCC: Hepatocellular carcinoma.
GWAS
Modulation of glucose metabolism
Induction of steatosis
PNPLA3 PPP1R3B TM6SF2 GCKR NCAN LYPLAL1
Oxidative stress
inflammation fibrogenesis
PNPLA3TM6SF2GCKRFDFT1PDGFA
COL13A1LTBP3
EFCAB4B
PNPLA3Carcinogenesis
Inflammation and fibrosis
Cirrhosis and HCC
Steatosis
Candidate genes studies
LEPRRETNPEMTFATP5ADRB2ADRB3PPARa
PPARGC1APPARγAPOEAPOC3MTTPLPIN1
ADIPOQ ENNP1IRS-1
TNF-aTRAILIL-6IL-1bTLR4IL28BSOD2
CYP2E1UCP3UCP2
MTHFRGCLCHFE
TMPRSS6KLF6
TGF-β1ATⅡ
AGTR1
Macaluso FS et al . Genetic background in NAFLD
Normal liver
NAFLD - ALD - DAFALD
PNPLA3rs738409
16 g/d
24 g/d 80 g/d
60 g/d
40 g/d
24 g/d
ALDNASH
PNPLA3rs738409

PNPLA3 in alcoholic liver diseasePNPLA3 rs738409 n Fenotipo
Trepo et al 2011 330 ASH + cirrhosis
Tian et al 2010 1221 ASH + cirrhosis
Seth et al. 2010 548 ASH + cirrhosis
Stickel et al 2011 1043 ASH + cirrhosis
Trepo E et al. J Hepatol 2011;55:906-12Trepo E et al. Hepatology 2012;55:1307-8.Tian et al. Nat Genet 2010;42:21-3.Stickel et al. Hepatology 2011;53:86-95.Stickel, F et al. J Hepatol 2017
Allele GGenotype GG
steatosisNecroinflammation
Fibrosis
CirrhosisHCC
locations with the greatest evidence for linkage withalcohol dependence contain several plausible candi-dates such as the ADH and GABA receptor gene clus-ters, both of which are on chromosome 4.
Genome-wide association studies. Genome-wideassociation studies (GWAS) entail the extensive par-allel genotyping of hundreds of thousands of geno-mic markers, typically SNPs, which cover themajority of common genetic variation across thehuman genome. GWAS are performed in large, gen-erally unrelated, populations in which either quali-tative or quantitative phenotypic data areavailable. Genetic association is identified when anallele or genotype is associated with a phenotypeat a specific significance threshold, which takes intoaccount the need for multiple testing, and is typi-cally set at p <5 ! 10"8. Independent verificationthrough replication analysis in a separate popula-tion or cohort is then advised. It follows that largepopulations are required in order to ensure thatGWAS are adequately powered. These studies arehypothesis-generating in that they are not basedon a priori hypotheses.
Several GWAS of alcohol dependence and associ-ated phenotypes have been undertaken [86–96], alarge proportion of which are based on collaborativestudies in the USA (Table 5). In most instances thesecohorts are phenotypically heterogeneous, contain-ing participants of multiple different, or mixed,ancestries, a high proportion of whom have co-morbid psychiatric disorders and/or co-occurringdrug dependence. Many of the GWAS undertakento date have failed to identify significant genome-wide associations. However, meta-analyses andstudies in populations with greater phenotypic sur-ety have identified significant genome-wide associ-ations between variants in the genes responsiblefor alcohol metabolism, e.g., ALDH1B and ALDH2 inEast Asian ancestry populations [69,74] and ADH1Band ADH1C in European, African and East Asianancestry populations [69,73]. Other significant asso-ciations, when identified, appear to be specific toindividual studies. However, in a recent minorallele-based meta-analysis of four GWAS, multiplegenes with significant or suggestive association withalcohol dependence were identified, some of whichare supported by evidence from linkage and candi-date gene studies [97].
activity resulting in the production of excess acetaldehyde. It isfound in 19–91% of East Asians [69] but in 0–10% of otherpopulations [70]. (C) The SNP rs671 in ALDH2 results in a loss offunction and hence a decrease in enzyme activity leading to theaccumulation of acetaldehyde. It is found in 30 to 50% of EastAsians and is almost exclusively confined to these populations[71].
Rev
iew
Acetaldehyde ALDHADHAlcohol
NAD+ NADH + H+ NAD+ NADH + H+
Acetate
A
Acetaldehyde ALDHADHAlcohol
NAD+ NADH + H+ NAD+ NADH + H+
Acetate
rs1229984ADH1B
B
Acetaldehyde ALDHADHAlcohol
NAD+ NADH + H+ NAD+ NADH + H+
Acetate
rs671ALDH2
C
Fig. 1. The effects of functional variants in the genes encoding the alcohol metabolizing enzymesalcohol dehydrogenase and acetaldehyde dehydrogenase. (A) Alcohol is metabolized in the liverprimarily by alcohol dehydrogenase (ADH) and acetaldehyde dehydrogenase (ALDH). Functionalvariants in the genes encoding the alcohol metabolizing enzymes are associated with changes inenzyme kinetics which affect the production and removal of the toxic metabolite acetaldehyderesulting in an increase in it circulating levels. The physical consequences of this act as a deterrent todrinking and hence ‘protection’ against alcohol use disorders and their sequelae. (B) The single nuclearpolymorphism (SNP) rs1229984 in ADH1B results in a gain of function and hence an increase in enzyme
Review
200 Journal of Hepatology 2017 vol. 66 j 195–211
Future directions
Alcohol has wide-spread adverse effects on a num-ber of neurobiological systems. However, theeffects of the genetic risk variants for alcoholdependence identified so far are small. It is highlyunlikely that the inheritance of harmful drinkingand alcohol dependence is simply controlled. It ismore likely to be polygenic and complex as it prob-ably involves transmission of one or more interme-diate characteristics or endophenotypes whichsubsequently affect the risk for harmful drinkingand dependence. Each of these endophenotypes is
likely to reflect the actions of multiple genes andto reflect both genetic and environmental influ-ences. Future studies should take advantage ofimprovements in technology; use large populationconsortia; and, avoid population heterogeneity andthe confounding effects of co-morbid and co-occurring disorders [36].
Alcohol-related liver disease
ALD is a term which encompasses a continuum ofpartly overlapping liver abnormalities ranging from
Rev
iew
Table 5. Genome-wide association studies of alcohol use phenotypes.
First author and date [Ref.]
Location Cohort Ethnicity Drinking behaviour Cases Controls Genes with p value ≤5 x10-8N Sex N Sex
Treutlein, 2009 [86] Germany - European Alcohol dependence 1151 Male 2354 Male PECRBierut, 2010 [87] USA/Germany SAGE African American
European AmericanAlcohol dependence 1897 Both 1932 Both -
Edenberg, 2010 [88] USA COGA African American European American
Alcohol dependence 1192 Both 692 Both -
Lind, 2010 [89] Holland/Australia NESDA OZALC
European Alcohol dependence 1823 Both 2763 Both -
Heath, 2011 [90] Australia OZALC European Alcohol use disorder 2062 Both 3393 Both -Schumann, 2011 [91] Pan-European AlcGen European Alcohol consumption 47,501 Both - - AUTS2Bail, 2011 [92] South Korea - East Asian Alcohol consumption 2834 Male - - ALDH2Zuo, 2012 [93] USA SAGE
COGAAfrican American European American
Alcohol dependence 2090 Both 2016 Both KIAA0040
Frank, 2012 [94] Germany - European Alcohol dependence 1333 Male 2168 Males ADH1B-ADH1C Park, 2013 [95] South Korea - East Asian Alcohol dependence 621 Both 750 Both ADH1B, ALDH2 Gelernter, 2014 [96] USA GCD
SAGEAfrican American European American
Alcohol dependence 7677 Both 6992 Both ADH1B, ADH1C, LOC100507053 METAP, PDLIM5
USA, United States of America; SAGE, Study of Addiction: Genetics and Environment; COGA, Collaborative Study on the Genetics of Alcoholism; NESDA Netherlands Study ofDepression and Anxiety; OZALC, Australian Twin-Family Study of Alcohol Use Disorder; AlcGen, Alcohol-GWAS consortium; GCD, GWAS discovery samples; PERC, per-oxisomal trans-2-enoyl-coA reductase; AUTS2, autism susceptibility candidate 2 gene; ALDH, acetaldehyde dehydrogenase; ADH, alcohol dehydrogenase; METAP, methionylaminopeptidase; PDLIM5, PDZ and LIM domain 5.
Table 4. Genome-wide linkages studies of alcohol use phenotypes.
First author and date [Ref.]
Location Cohort Families (n)
Ethnicity Drinking behaviour Region of interest LOD >3
Potential regional candidates
Reich, 1998 [77] USA - 105 European Alcohol dependence - -Long, 1998 [78] USA - 172 Native
AmericanAlcohol dependence Chromosome 4p
Chromosome 11p GABRB1 DRD4 and TH
Ehlers, 2004 [79] USA - 100 Native American
Alcohol dependence ≈ Chromosome 4p ADH1B
Wyszynski, 2003 [80] USA Framingham Heart Study
330 European Heavy alcohol consumption
- -
Wilhelmsen, 2005 [81] USA SMOFAM 158 European Alcohol dependence - -Prescott, 2006 [82] Ireland IASPSAD 474 European Alcohol dependence/
alcohol misuse Chromosome 4 q22 to q32
ADH cluster
Gelernter, 2009 [83] USA - 238 African America
Alcohol dependence Chromosome 10 q23.3 to q24.1
-
Hansell, 2010 [84] Australia - 1690 European Alcohol dependence - -Gizer, 2011 [85] USA UCSF Family
Alcoholism Study713 European Alcohol dependence - -
USA, United States of America; SMOFAM, Smoking in Families Study; IASPSAD, Irish Affected Sib-Pair Study of Alcohol Dependence; UCSF, University of California SanFrancisco; DRD4, dopamine receptor D4; TH, tyrosine hydroxylase; GABRB1, gamma-aminobutyric acid (GABA) A receptor beta 1; ADH, alcohol dehydrogenase; LOD,Logarithm of the odds.
JOURNAL OF HEPATOLOGY
Journal of Hepatology 2017 vol. 66 j 195–211 201

PNPLA3 rs738409 & TM6SF2 rs58542926allele G and allele T increased risk of NAFLD
MAF AFR AMR ASIA EUR
PNPLA3 –G-‐ 0.12 0.48 0.35 0.23
TM6SF2 –T-‐ 0.02 0.06 0.09 0.07Allele frequencies information was collected from 1000 Genomes Project (Genomes Project et al., 2015). AFR, African; AMR, American; EAS, East Asian; EUR, European.
Wang et al. Front Genet 2016;7:140
N=367 NAFLDN= 366 Health
122/291 191/365 63/102 N=240 n=336 n=135 n=12
P<0.001 P<0.001

TM6SF2 and MBOAT7 and liver phenotype
Eslam M, et al. Hepatology 2016; Mancina et al. Gastro 2016
MBO
AT7
TM6S
F2

NAFLD: PNPLA3, MBOAT7, TM6SF2
28
Table 6 Risk factors for developing hepatic steatosis
(A) Univariate analysis
Factor OR 95% CI P
PNPLA3 p.I148M 2.418 1.323 – 4.419 0.004
MBOAT7 rs641738 1.260 0.749 – 2.119 0.384
TM6SF2 p.E167K 4.622 1.077 – 19.831 0.039
Glucose 1.015 0.994 – 1.037 0.168
BMI 0.966 0.933 – 1.001 0.055
Age (years) 1.005 0.979 – 1.033 0.692
Sex 2.080 0.933 – 4.634 0.073
Presence of diabetes 1.224 0.504 – 2.973 0.656
Triglycerides 1.002 0.996 – 1.007 0.594
Cholesterol 0.997 0.988 – 1.007 0.539
(B) Multivariate analysis
Factor OR 95% CI P
PNPLA3 p.I148M 2.424 1.326 – 4.419 0.004
TM6SF2 p.E167K 4.725 1.093 – 20.429 0.038
Abbreviations: CI, confidence interval; E, glutamic acid; I, isoleucine; K, lysine; M, methionine; MBOAT7, membrane bound O-acyltransferase domain containing 7; OR, odds ratio; p, protein (amino acid number); PNPLA3, adiponutrin; TM6SF2, transmembrane 6 superfamily member 2. The relationship between steatosis PNPLA3, TM6SF2 and MBOAT7 variants as well as other potentially prosteatotic factors were assessed by uni- and multivariate logistic regression analysis. Genetic analyses were calculated by using either additive (for PNPLA3 and MBOAT7) or dominant (for TM6SF2) models.
by guest, on Novem
ber 14, 2016w
ww
.jlr.orgD
ownloaded from
29
Table 7 Risk factors for developing hepatic fibrosis
(A) Univariate analysis
Factor OR 95% CI P
PNPLA3 p.I148M 1.679 1.192 – 2.367 0.003
MBOAT7 rs641738 1.410 1.003 – 1.982 0.048
TM6SF2 p.E167K 1.060 0.587 – 1.914 0.846
Glucose 1.020 1.008 – 1.033 0.002
BMI 0.989 0.965 – 1.015 0.413
Age (years) 1.020 1.002 – 1.039 0.027
Sex 1.088 0.671 – 1.763 0.732
Presence of diabetes 2.092 1.136 – 3.852 0.018
Triglycerides 1.003 1.000 – 1.007 0.083
Cholesterol 0.997 0.991 – 1.003 0.314
(B) Multivariate analysis
Factor OR 95% CI P
PNPLA3 p.I148M 1.676 1.019 – 2.757 0.042
MBOAT7 rs641738 1.766 1.089 – 2.864 0.021
Abbreviations: CI, confidence interval; E, glutamic acid; I, isoleucine; K, lysine; M, methionine; MBOAT7, membrane bound O-acyltransferase domain containing 7; OR, odds ratio; p, protein (amino acid number); PNPLA3, adiponutrin; TM6SF2, transmembrane 6 superfamily member 2 The relationship between steatosis PNPLA3, TM6SF2 and MBOAT7 variants as well as other potentially profibrotic factors were assessed by uni- and multivariate logistic regression analysis. Genetic analyses were calculated using either additive (for PNPLA3 and MBOAT7) and dominant (for TM6SF2) models.
by guest, on Novem
ber 14, 2016w
ww
.jlr.orgD
ownloaded from
Risk factors for developing hepatic fibrosis
Risk factors for developing hepatic steatosis
Krawczyk, M et al. J Lipid Res 2016
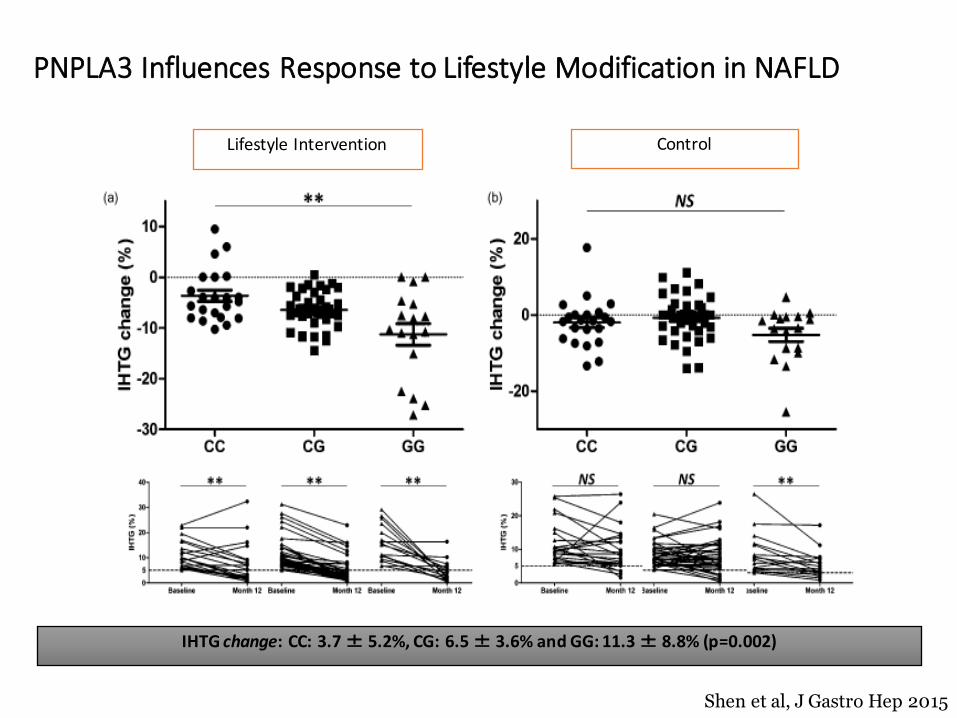
PNPLA3 Influences Response to Lifestyle Modification in NAFLD
Shen et al, J Gastro Hep 2015
IHTG change: CC: 3.7 ± 5.2%, CG: 6.5 ± 3.6% and GG: 11.3 ± 8.8% (p=0.002)
Lifestyle Intervention Control

AVE FIBROSIS RVSHLADRB1*01 HLA DRB1*11 SLC11A1IL6 (-6 C/C) HLA DRB1*03 HLADQB1*0301CCR5-D32 TGF-b1 TNFRANTES TNF NRAMP2
TfR1 CCR5 MxANRAMP2 RANTES PKRSLC11A1 HFE 5´2-OAS
KIR2DL3-HLAC1 MCP-2 20210PTSLC11A1 HLA-B44
APO-E TGF-b1NRAMP2 APO-E
FVL CCR5

SpontaneousViral
clerarance
Sustained Virological response
Fibrosis progression
N=1002 gen 2&3
Montes-Cano Hepatology 2010;; Eslam M et al J Hepat 2014;; Eslam Nat Comm 2015
IL28B polymorphism in Hepatitis C HCV HCV
infection
Chronic Hepatitis C
Spontaneous viral Clearance
Female IL28B-‐CC
Genotype 1Selection of just
one clone
BTN3A2
HCV (N=3129)

Hepatitis C rs12979860 [IFNL4]Relapses ION-‐3 (20 / 423) ION-‐4 (10 / 322) ION-‐3 + ION-‐4
IFNL4 rs1297986 Relapse p-‐value Relapse p-‐value RR 95% CI p-‐value
CC 1.8% 0.0%
CT 4.9% 1.6% 2.74 0.74-‐10.17
TT 9.1% 0.03% 10.3% 0.001 4.58 1.26-‐16.56 0.0002
Proportions of patients who suffered virological relapse in response to treatment with ledipasvir/sofosbuvir, by IFNL4 rs12979860 genotype.
10.2217/pgs.16.28 Pharmacogenomics (Epub ahead of print)
Figure 3. Ribavirin-induced anemia depending on the activity of ITPA.
Healthy erythrocyte Sick erythrocyte
ATPATP
ITPITP
GTP
ITPase ITPase
Deficient ITPA activity Normal ITPA activity
Membrane oxidative stress
future science group
Review Ampuero & Romero-Gómez
decline. Interestingly, the relationship between Hb decline and SVR was independent of ITPA geno-type [43]. In addition, ITPA genetic variants have been associated with severe RBV-induced anemia and could influence the efficacy of pegIFN and RBV in elderly patients harboring favorable IL28B [44]. Role of ITPA has been also established in HCV genotypes 2 and 3, decreasing the risk of anemia and reducing relapse rates in those patients showing reduced ITPase activ-ity [45]. Regarding predictive values, positive predic-tive value for ITPA has been demonstrated in 76% for anemia after 4 week of therapy and 91% for anemia at any time of the therapy, while negative predictive value was 61 and 28%, respectively [46]. Further in dual therapy, ITPA has been studied in telaprevir- and boceprevir-based therapies. ITPA rs1127354 CC geno-type influenced on the appearance of severe anemia (Hb <8.5 g/dl) in a Japanese cohort (n = 292) treated with telaprevir [47]. Previously, another Japanese study (n = 94) showed that the same polymorphism required RBV dose reduction earlier in patients with genotype 1 treated with PegIFN, RBV and telaprevir [48]. How-ever, a limited effect of ITPA polymorphisms has been observed in patients with advanced liver fibrosis receiv-ing telaprevir [49]. In Caucasian population, those show-ing reduced ITPase activity had a higher Hb level after 4 weeks of treatment either with telaprevir or bocepre-vir compared with patients with normal ITPase activ-ity [50]. In addition, normal ITPase activity has been independently associated with anemia in 687 patients treated with boceprevir, increasing two-times the risk of Hb <10 g/dl [51]. New drugs, such as simeprevir,
cause fewer anemia rates than first-generation protease inhibitors [52]. Regarding to this drug, ITPA polymor-phism (rs1127354) influenced Hb levels and incidence of RVB dose reduction during simeprevir triple therapy in 212 patients with genotype 1 [53]. We have no data about the rest of new DAAs, but probably we should be cautious with elder anemic patients.
SLC28/29 genesSLC28A2 and SLC28A3, as well as SLC29A1 and SLC29A2 have been evaluated in some studies, due to the potential interest in RBV-induced anemia. First study was performed in a German cohort (n = 169, HCV genotype 1) treated with dual therapy. They were genotyped for 21 variants in SLC28A2, SLC28A3, SLC29A1 and SLC29A2 genes and observed that SLC28A3 haplotype rs10868138G/rs56350726T was associated with a lower incidence of Hb drop >3 g/dl [54]. In a Swiss cohort, 216 patients (61% geno-type 1) were treated with pegIFN-RBV and RBV serum levels were obtained in some of them (n = 67). They found that SLC28A2 rs11854484 TT genotype had higher body weight-adjusted RBV levels and showed a trend toward anemia, while SLC28A3 rs56350726 and SLC28A3 rs10868138 were not associated with anemia, Hb drop or RBV serum levels. The conclu-sion was that SLC28A2 rs11854484 TT genotype could increase the RBV absorption and, consequently, cause anemia. Authors also evaluated the combination between SLC28 and ITPA, but no potential interest could be observed [55]. Regarding protease inhibitors-based therapy, there are a few studies assessing the
Ampuero, J et al. Pharmacogenomics 2016; Morio, K. et al. J Gastroenterol 2016; O’Brien, T et al. Hepatology 2017
ITPA rs1127354 C/ASCL28A2 rs1127354 C/T

HCV (N=3129)
Eslam et al, Nat Commun. 2015;6:6422
Interferon-λ rs12979860 genotype and liver fibrosis progression

Association between fibrosis progressionrate and combining risk analysis (FPR score = PNPLA3 + IFNL4)
Tamaki N, et al PLOS ONE 2015 10(9): e0137351.
PNPLA3: Allele GIFNL4: Allele C

IL28B CC easier to cure faster progression
International Liver Disease Genetics Consortium (ILDGC). FibroGENE: A gene-based model for staging liver fibrosis. J Hepatol. 2016;64(2):390-8.

FibroGENE-DT: clinical utility
These findings could have immediate clinical utility for:ü The exclusion of cirrhosis in subjects with CHC, CHB and NAFLD, negative predictive value (NPV) >0.96.
ü Identifying those with a high likelihood of advanced fibrosis in CHC, CHB and NAFLD.
ü Patient counseling on the risk of rapid fibrosis progression in CHC.
FibroGENE: Systematic un-biased utilizing of non-parametric, machine learning methods

Viral Hepatitis Bà HLA-DPA1à HLA-DPB1à NTCP
rs3077 rs9277535rs2296651
- Spontaneous viral clearance- Progression to liver cancer
(25fold).- Response to interferon.
Chih, L et al. CHM 2016

IFNL4 & MBOAT7 and Fibrosis in hepatitis B
N=555
A B
Supplementary figure 1: Association of MBOAT7 rs641738 genotype with inflammation degree and fibrosis stage in the population
(n=1101) using the dominant model for the minor allele.
Page 35 of 46
Hepatology
Hepatology
5051525354555657585960
This article is protected by copyright. All rights reserved.
MBOAT7 rs641738
Thabet, K et al. Hepatology 2017; Eslam et al. Nat commun 2015

GWAS in PBC: IL-12 a case report
Ustekinumab, a monoclonal antibody which targets the
p40 subunit of IL-12 and IL-23, tested in PBC, demonstrated lack
of efficacy (https://clinicaltrials.gov/ct2/
show/NCT01389973)
Gulamhusein AF et al. Sem Liv Dis 2015;35:292

Autoimmune Liver DiseasesPBC/AIH/PSC
Karlsen et al. J Hepatol 2015
Diseases caused by distinct exogenous factors (e.g., drug-induced liver injury, AIH, PBC, PSC, celiac disease, or Crohn Disease showed a similar genetic architecture, where a strong association with genes in the major histocompatibility complex (MHC, plotted in red) is the defining feature.

HLA-DRB1*1501-DQB1*0602 (classII)HLA –A*0201 (class I)
Lucena et al, Gastroenterology 2011; Stephens et al, Plos One 2013
GWAS analysis in DILI
HLA associations in Spanish population (Amoxicillin-DILI n=75 vs. n=885 healthy controls)

DILI susceptibility risk factors
ü Many genetic variants previously related to DILI were not confirmed in GWAS analysis.
ü All GWAS analysis demonstrated HLA role in DILI risk. ü Statistical power and association of genetic variants were weak. ü As a complex disease multiples genetic variants should play together to pormote
DILI.

HE in our genes: a microsatellite in the promoter region of glutaminase gene.
Romero-Gómez et al. Ann Intern Med 2010;; Mayer et al. J Int Med 2015
HEPATIC ENCEPHALOPATHY
Frequency distribution of MS16xGCA alleles in the Spanish population (n=768
chromosomes)
GCA Repeat number30,0025,0020,0015,0010,005,00
Frecuency
300
200
100
0
Long-Long microsatellite increases x3 HE bouts
Luciferase reporter activity according to the length of microsatellite allele
This was reflected by a significant difference in CFFscores in patients with these alleles, compared toshort and short–long form allele carriers. Further-more, multiple regression analysis demonstratedthat CFF values were independently influenced bythe GLS variant. Evidence of a genetic risk for HEwas first provided by Romero-Gomez et al. [26] whoobserved an association between HE incidence andthe GLS microsatellite in a Spanish cohort. Theauthors also reported that the long form of the GLSmicrosatellite correlated with increased GLS activ-ity in vivo. A similar proportion of patients with thegenetic risk variant were identified here and in theSpanish cohort (~30%) despite the different meth-odologies used in the two studies (i.e. capillaryelectrophoresis by Romero-Gomez et al. [26] andboth gel and capillary electrophoresis herein).Consequently, the precise definitions of the alleleswere not identical. Moreover, different thresholdsof CFF values for diagnosing the presence of HEwere used. In the present study, HE was diagnosedat a test frequency of ≤39 Hz, compared to ≤38 Hzin the study of Romero-Gomez et al. [26]. Never-
theless, despite these differences, an associationbetween the GLS promoter polymorphism and thedevelopment of HE was replicated in the presentstudy. Moreover, a larger proportion of patientswith overt HE in the presence of two long alleles inthe GLS microsatellite promoter region wasobserved in both studies.
Taken together, our findings support the notionthat intestinal GLS is a risk gene for HE. Theintestine has been implicated as an importantsource of ammonia production due to raised GLSactivity, as seen in patients with cirrhosis [22]. Theresults from oral glutamine challenge tests havesupported the view that ammonia produced by thesmall intestine contributes to the development ofHE [30, 31]. Romero-Gomez and co-workers founda strong correlation between GLS activity andminimal HE [22], and lower glutamine levels havealso been detected in patients with HE [2]. Conse-quently, the excess glutamate and ensuing excit-atory neurotransmitters might promote HEsymptoms. Although hyperammonaemia in HE iscommon, normal ammonia concentrations inpatients with HE have also been reported [1].Elevated ammonia levels were detected in one-third of patients in the present study and did not
Fig. 1 Proportion of patients with and without hepaticencephalopathy (HE) based on glutaminase (GLS) micro-satellite grouping. Significantly more patients with theGLS long form presented with HE, compared to those withthe short or short–long forms (v2 = 5.5, P = 0.019). Thepresence and severity of HE were assessed using thecritical flicker frequency test (≤39 Hz denotes HE cases).
Table 3 Logistic regression analysis of HE predictors
Factor OR 95% CI P
(A) Univariate analysis with the presence of HE as
dependent variable
Age 1.04 1.01–1.06 0.012
GLS long form 2.28 1.14–4.57 0.020
GLS short–long form 0.55 0.29–1.03 0.062
Sodium (low vs. normal)a 2.52 1.21–5.27 0.014
Liver cirrhosis aetiology
(alcoholic vs. non-alcoholic)
2.00 1.05–3.80 0.035
SBP 3.00 1.03–8.71 0.043
(B) Multivariate analysis with the presence of HE as
dependent variable
Age 1.04 1.01–1.07 0.016
GLS long form 3.23 1.46–7.13 0.004
Sodium (low vs. normal)a 2.96 1.32–6.61 0.008
Liver cirrhosis aetiology
(alcoholic vs. non-alcoholic)
2.13 1.04–4.35 0.039
CI, confidence interval; OR, odds ratio; SBP, spontaneousbacterial peritonitis; HE, hepatic encephalopathy. aLowsodium levels (<135 mmol L!1) versus high sodium levels(>135 mmol L!1).
L. B. Mayer et al. GLS gene in hepatic encephalopathy
ª 2015 The Association for the Publication of the Journal of Internal Medicine 319
Journal of Internal Medicine, 2015, 278; 313–322

ACUTE ON CHRONIC LIVER FAILURE
257x190mm (300 x 300 DPI)
Page 38 of 61
Hepatology
Hepatology
This article is protected by copyright. All rights reserved.
191x254mm (300 x 300 DPI)
Page 39 of 61
Hepatology
Hepatology
This article is protected by copyright. All rights reserved.
191x254mm (300 x 300 DPI)
Page 39 of 61
Hepatology
Hepatology
This article is protected by copyright. All rights reserved.
Alcaraz-Quiles et al. Hepatology 2017

PNPLA3 rs738409
SUSCEPTIBILITY
PHAR
MAC
O-
GEN
OM
ICS
We do not have enough information yet to implement most of the new data in clinical practice

Ampuero et al. Pharmacogenomis 2016Eslam M et al. Semin Liver Dis, 2015;35:402-420.
ExploreEpigenetics
lncRNA, miRNAs
CONCLUSIONES:La genética de las enfermedades hepáticas se regulan por tres principios:1.- VEROSIMILITUD2.- FUNCIONALIDAD3.- UTILIDAD CLÍNICA








