

Fibrosis Quística Ehler DanlosOsteogenesis Imperfecta
Fatima Garcia 11611300Jissela Peralta 11611051Maria Fernanda Cruz 11611145

FIBROSIS QUISTICA

Fibrosis Quística• Historia: En 1936, el pediatra suizo Guido Fanconi
• Etiología: el defecto principal en la fibrosis quística se relaciona con una función anormal de una proteína de los canales del cloro epiteliales codificada por el gen del regulador de la conductancia transmembrana de la fibrosis quística (CFTR).
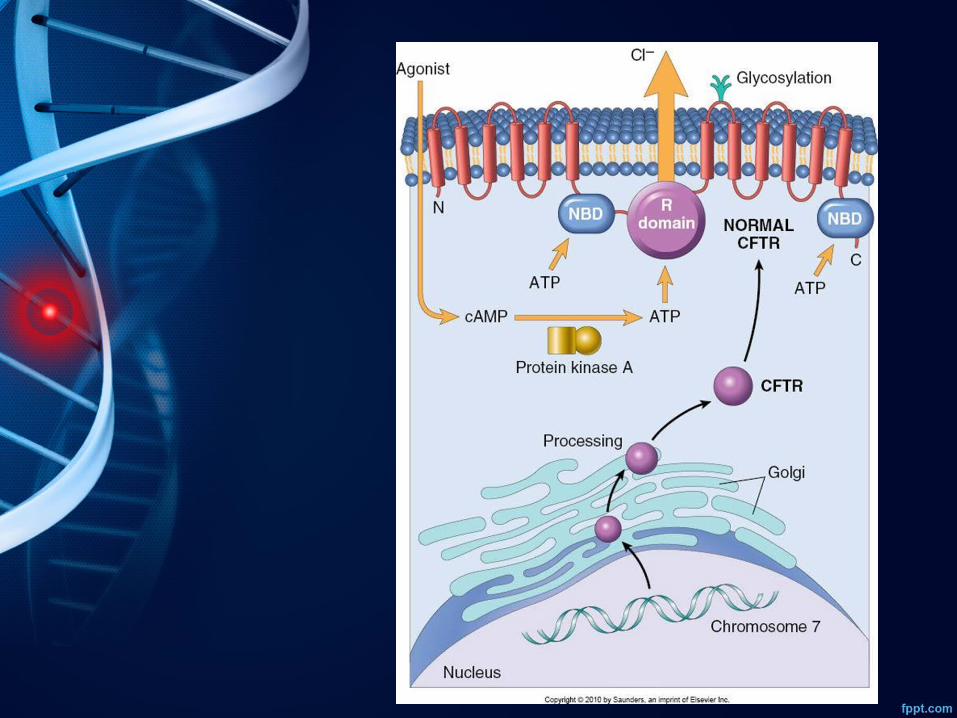


Incidencia • Es más frecuente en la población blanca no hispana• Estado Unidos, Europa y Australia es de 1 de cada
3,000-5,000 nacimientos. • Hispanos (1 de cada 7,000) • Afroamericanos (1 de cada 12,000)• Es rara en personas de origen asiático.

Factores de riesgo• Edad • Consumo de cigarrillo• Ocupación• Tratamiento de cáncer• Factores genéticos

Clasificación de Cromosopatía
• Es una enfermedad hereditaria mendeliana o monogénica.
• En la fibrosis quística el afectado es CFTR ubicado en el cromosoma 7q31.2
• Herencia: Autosómica recesiva

Tipos de Variante • CLASE I: síntesis defectuosa de la proteína• CLASE II: plegamiento, procesamiento y circulación
anormales de la proteína• CLASE IV: reducción de la conductancia• CLASE V: reducción de la abundancia• CLASE VI: alteraciones de la regulación de canales
iónicos separados
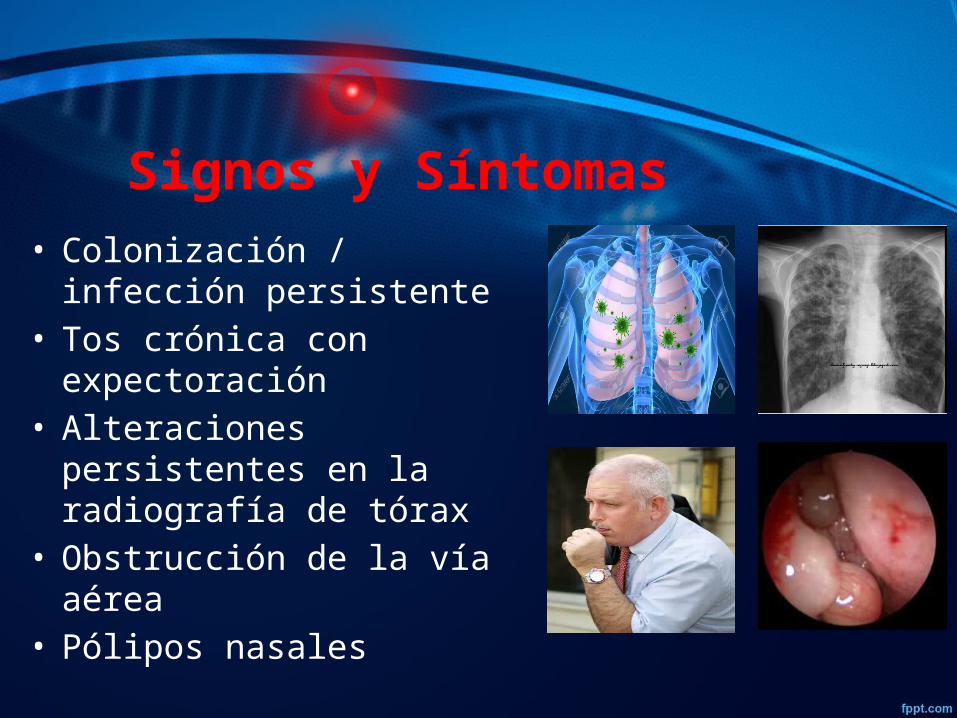
Signos y Síntomas • Colonización / infección
persistente • Tos crónica con
expectoración • Alteraciones persistentes en
la radiografía de tórax • Obstrucción de la vía aérea• Pólipos nasales

Diagnostico
Características fenotípicas:• Cuadro clínico compatible• Historia familiar• Tamizaje neonatal positivo • Ausencia bilateral de conductos deferentes • Disfunción del CFTR• Test de sudor positivo (en 2 ocasiones)• Estudio genético positivo (2 mutaciones
del CFTR)

Tratamiento • Tratamiento respiratorio • Tratamiento digestivo
nutricional • Terapia proteica • Terapia génica

Consejería GenéticaEl asesoramiento genético es el proceso a través del cual:• Se evalúan sus antecedentes médicos personales y los
de su familia• Se ayuda a los padres para que tomen decisiones
informadas sobre cómo proceder a continuación• Qué tan probable es que su hijo vaya a tener un
trastorno genético.
EHLER DANLOS

Ehler Danlos• Historia: Edward Ehler y Henry Alexander Danlos
(1991)
• Etiología: se debe a defectos de la síntesis de colágeno

Factores de Riesgo • Dolor articular crónico.• Artritis de aparición temprana.• Dificultad para cerrar las heridas
quirúrgicas o para retirar los puntos.• Ruptura prematura de membranas
durante el embarazo.• Ruptura de los grandes vasos
sanguíneos• Ruptura de un órgano hueco • Ruptura del globo ocular.

Tipos de Variante
• Clásico • Hiperlaxitud • Vascular • Cifoscoliosis• Artroclasia• Dermatopraxis

Ehler Danlos Tipo Clásico
• Locus: 2q32.2• Gen afectado: COL5A1, COL5A2• Herencia: autosómica dominante• Incidencia: 1/20,000 personas
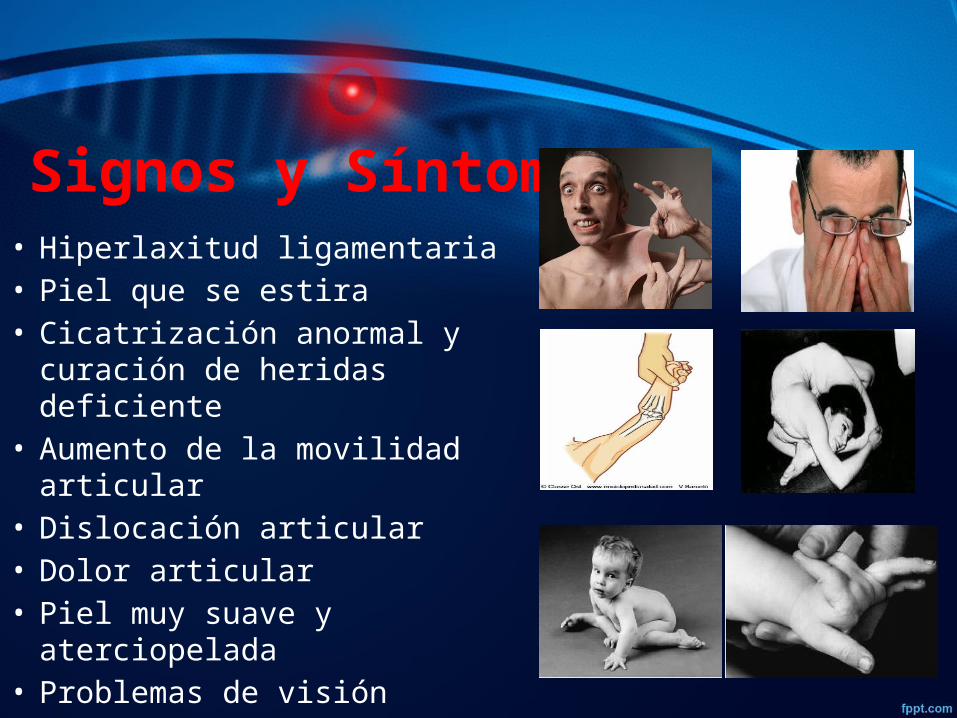
Signos y Síntomas • Hiperlaxitud ligamentaria• Piel que se estira• Cicatrización anormal y
curación de heridas deficiente• Aumento de la movilidad
articular• Dislocación articular• Dolor articular• Piel muy suave y aterciopelada• Problemas de visión

Ehler Danlos Tipo Hiperlaxitud
• Gen afectado: TNXB• Herencia: autosómica dominante• Incidencia: 1/5.000-20.000 personas
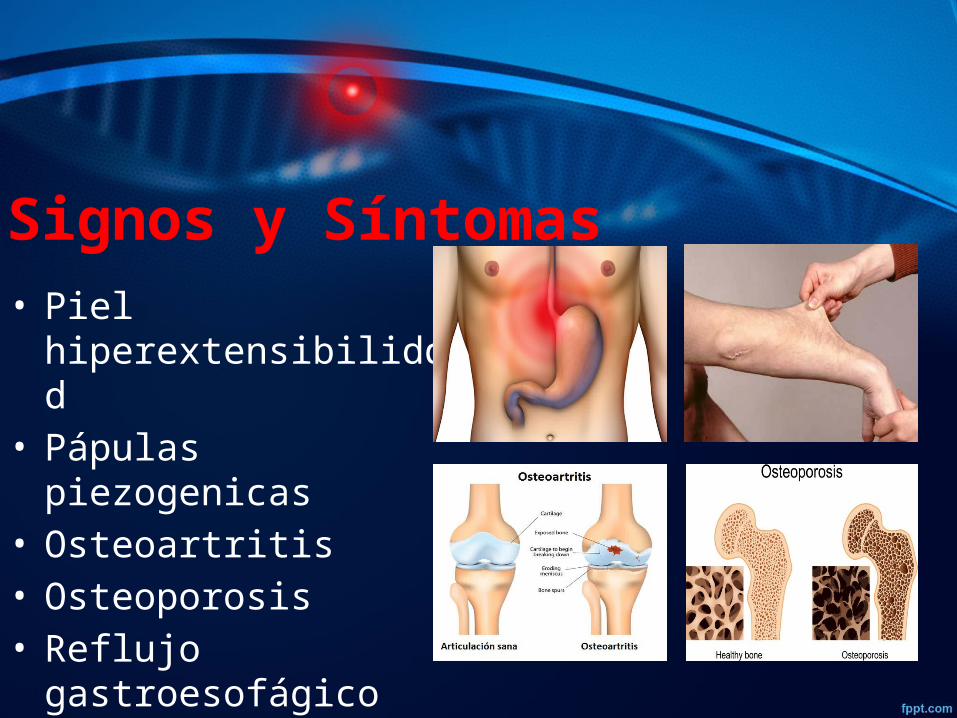
Signos y Síntomas • Piel hiperextensibiliddad• Pápulas piezogenicas• Osteoartritis• Osteoporosis• Reflujo gastroesofágico• Hiperlaxitud cardiaca

Ehler Danlos Tipo Vascular
• Gen afectado: COL3A1• Herencia: autosómica dominante• Incidencia: 1/50.000-250.000 personas.
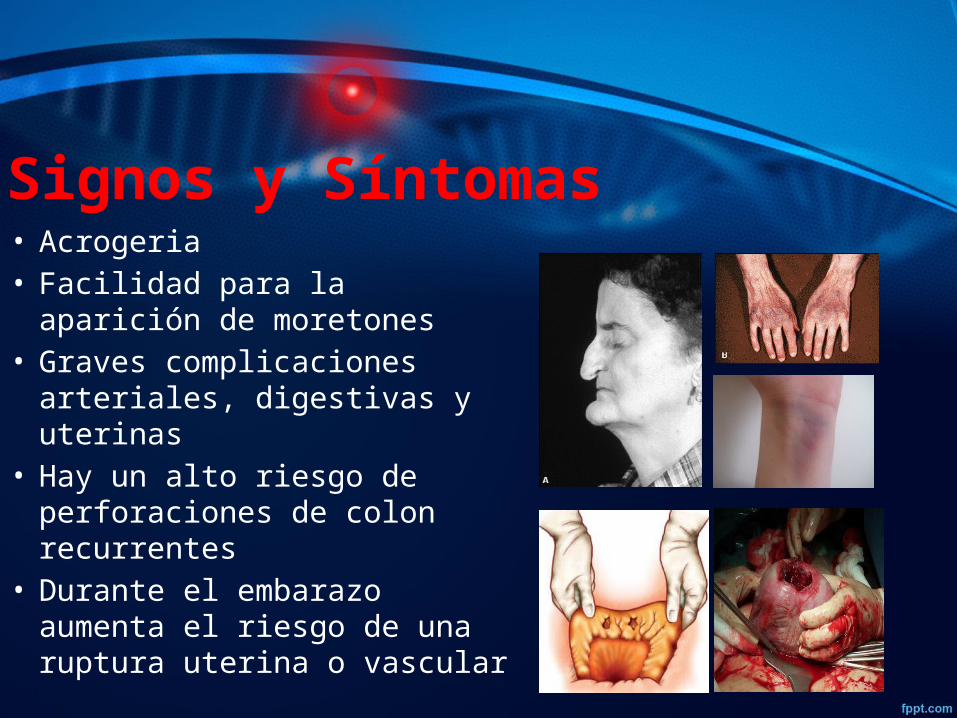
Signos y Síntomas• Acrogeria • Facilidad para la aparición de
moretones • Graves complicaciones
arteriales, digestivas y uterinas• Hay un alto riesgo de
perforaciones de colon recurrentes
• Durante el embarazo aumenta el riesgo de una ruptura uterina o vascular

Elher Danlos tipo Cifoscoliosis • Gen afectado: PLOD1• Herencia: Autosómica Recesiva• Incidencia: 1/100.000 personas.
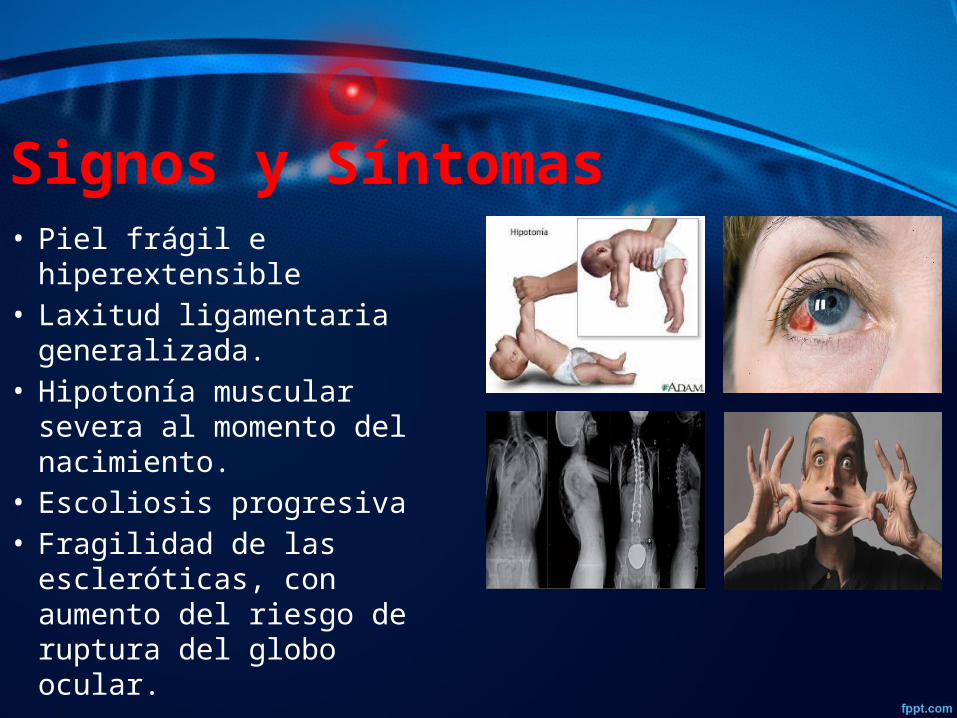
Signos y Síntomas• Piel frágil e hiperextensible • Laxitud ligamentaria
generalizada.• Hipotonía muscular severa
al momento del nacimiento.• Escoliosis progresiva• Fragilidad de las
escleróticas, con aumento del riesgo de ruptura del globo ocular.

Elher Danlos Tipo Artrocalasia
• Gen afectado: COL1A1, COL1A2• Herencia: Autosómica dominante• Incidencia: se ha descrito en 30 personas en el
mundo.
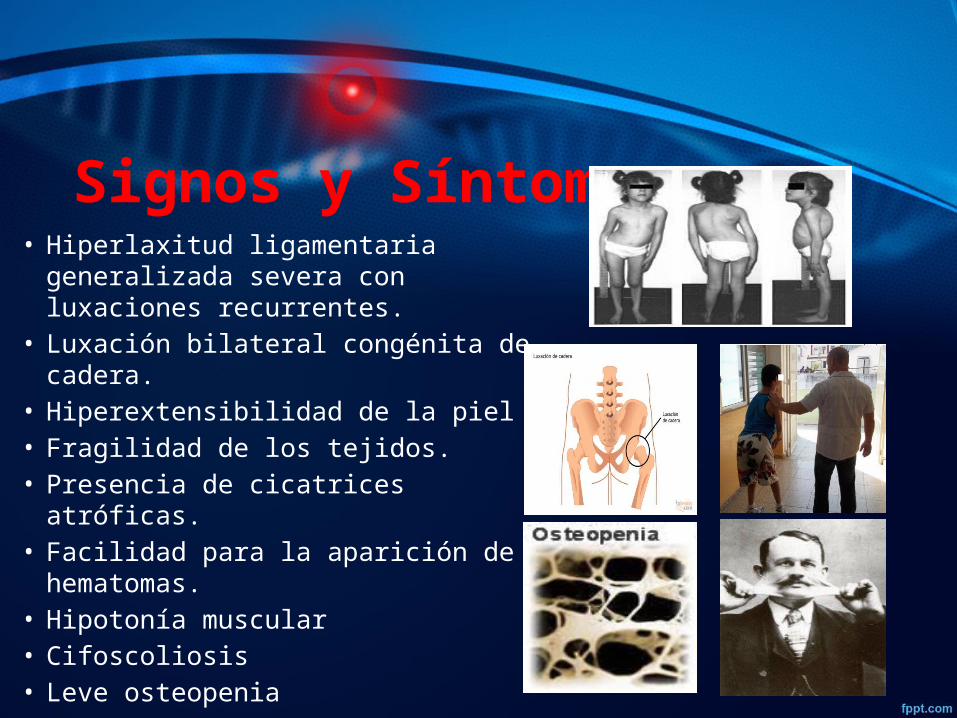
Signos y Síntomas• Hiperlaxitud ligamentaria generalizada
severa con luxaciones recurrentes.• Luxación bilateral congénita de cadera.• Hiperextensibilidad de la piel• Fragilidad de los tejidos.• Presencia de cicatrices atróficas.• Facilidad para la aparición de
hematomas.• Hipotonía muscular• Cifoscoliosis• Leve osteopenia

Ehler Danlos Tipo Dermatopraxis
• Gen afectado: ADAMTS2• Herencia: Autosómica recesiva• Incidencia: se ha descrito en 8 personas en el mundo.
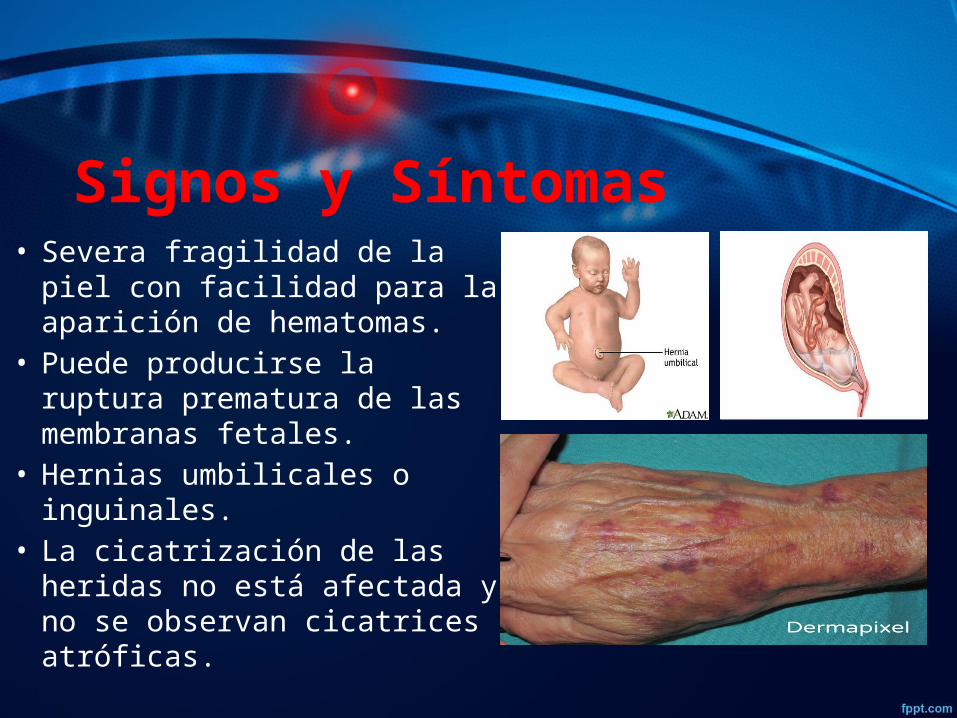
Signos y Síntomas• Severa fragilidad de la piel con
facilidad para la aparición de hematomas.
• Puede producirse la ruptura prematura de las membranas fetales.
• Hernias umbilicales o inguinales.
• La cicatrización de las heridas no está afectada y no se observan cicatrices atróficas.

Diagnostico • Tipificación del colágeno• Prueba de mutación del gen de
colágeno.• Ecocardiografía• Actividad de lisil oxidasa o de
lisil hidroxilasa

Tratamiento • Reposo de la articulación
afectada• Terapia ocupacional• Tomar de 0,4 a 1 mg de ácido
fólico diarios en forma permanente
• Tratamiento de la disautonomía • Tratamiento en equipo

Consejeria Genetica • Quienes estén planeando tener hijos deben ser
conscientes del tipo de síndrome de Ehlers-Danlos que tienen y la forma en que se transmite de padres a hijos.
Osteogenesis Imperfecta

Osteogenesis Imperfecta• Historia: Olof Ekman en su tesis doctoral (1788)
• Etiología: trastorno hereditario más frecuente del tejido conjuntivo de colágeno tipo 1
• Genes afectados: COL1A1 y COL1A2

Tipos de Variantes • Osteogenesis imperfecta tipo I Leve• Osteogenesis imperfecta tipo II Perinatal, letal• Osteogenesis imperfecta tipo III Deformante
progresiva • Osteogenesis imperfecta tipo IV Deformante con
escleróticas normales

Otras formas de OI que no dependen de la proteína del colágeno tipo 1 directamente se clasifican en nuevos grupos:• Osteogenesis Imperfecta tipo V• Osteogenesis Imperfecta tipo VI• Osteogenesis Imperfecta tipo VII• Osteogenesis Imperfecta tipo VIII• Osteogenesis Imperfecta tipo IX• Osteogenesis Imperfecta tipo X
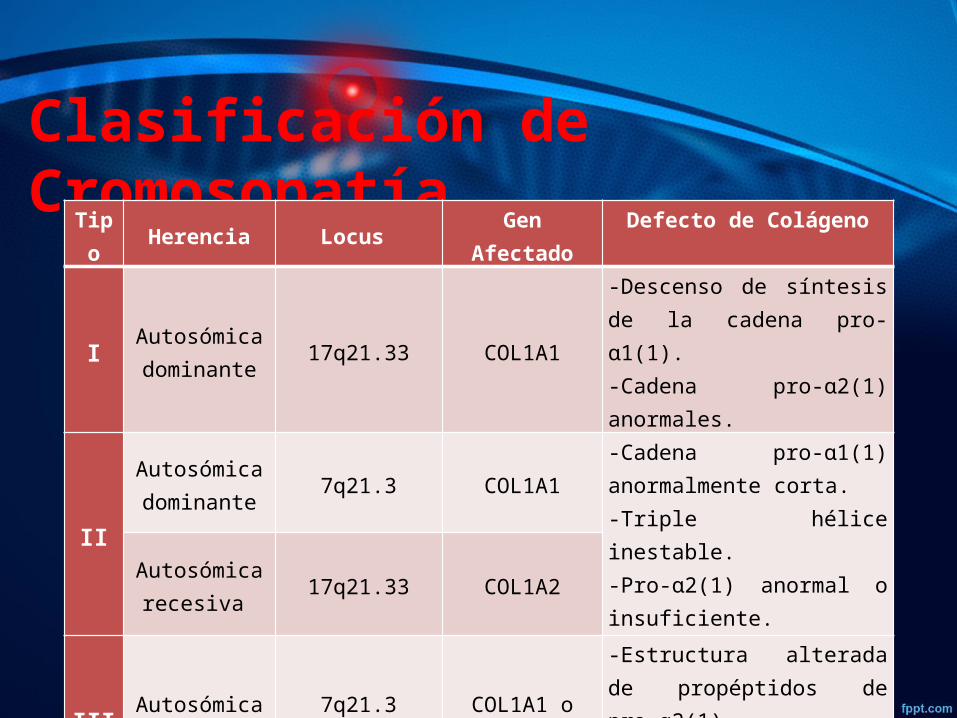
Clasificación de CromosopatíaTipo Herencia Locus Gen Afectado Defecto de Colágeno
I Autosómica dominante 17q21.33 COL1A1
-Descenso de síntesis de la cadena pro-ɑ1(1).-Cadena pro-ɑ2(1) anormales.
II
Autosómica dominante
7q21.3
COL1A1
-Cadena pro-ɑ1(1) anormalmente corta. -Triple hélice inestable.-Pro-ɑ2(1) anormal o insuficiente.
Autosómica recesiva 17q21.33 COL1A2
III Autosómica dominante
7q21.317q21.33
COL1A1 o COL1A2
-Estructura alterada de propéptidos de pro-ɑ2(1).-Formación alterada de triple hélice.
IV Autosómica dominante
7q21.317q21.33
COL1A1 o COLIA2
-Cadena pro-ɑ2(1) anormales.-Triple hélice inestable.

Tipo de Herencia• La Osteogénesis Imperfecta presenta herencia
genética dominante
• En casos como la osteogenesis imperfecta tipo II existe una herencia autosómica recesiva.
Incidencia • Su incidencia se estima entre 1:10.000 y 1:15.000• Dentro de cada tipo de osteogenesis imperfecta más
comunes, tiene diferentes porcentajes de incidencia, esta son:
• Tipo I 70%• Tipo II 2%• Tipo III 20%• Tipo IV 5%
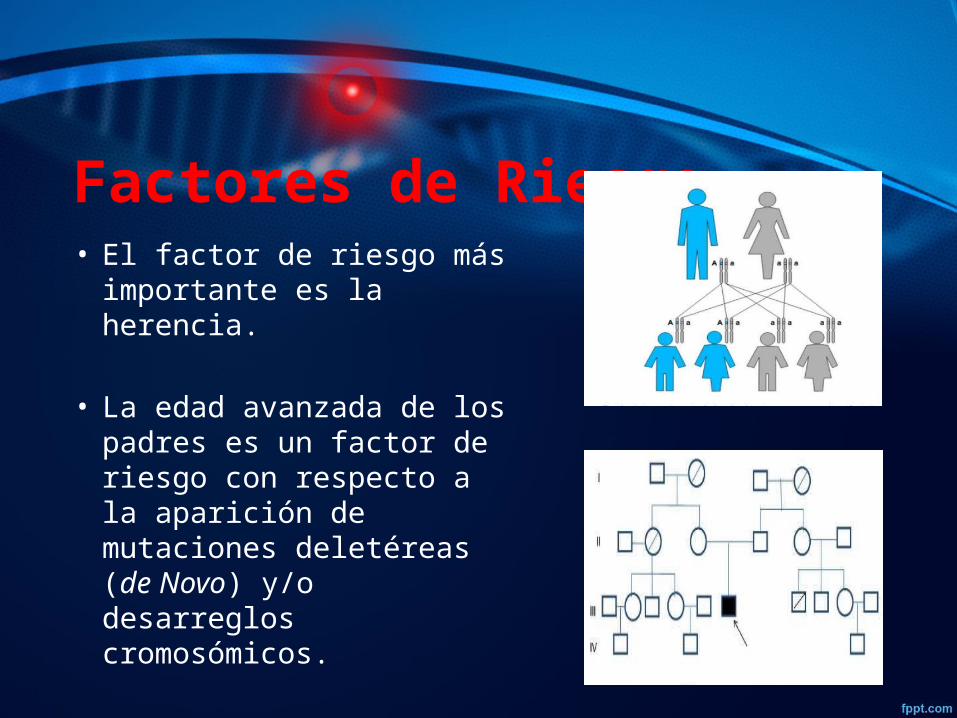
Factores de Riesgo • El factor de riesgo más
importante es la herencia. • La edad avanzada de los
padres es un factor de riesgo con respecto a la aparición de mutaciones deletéreas (de Novo) y/o desarreglos cromosómicos.
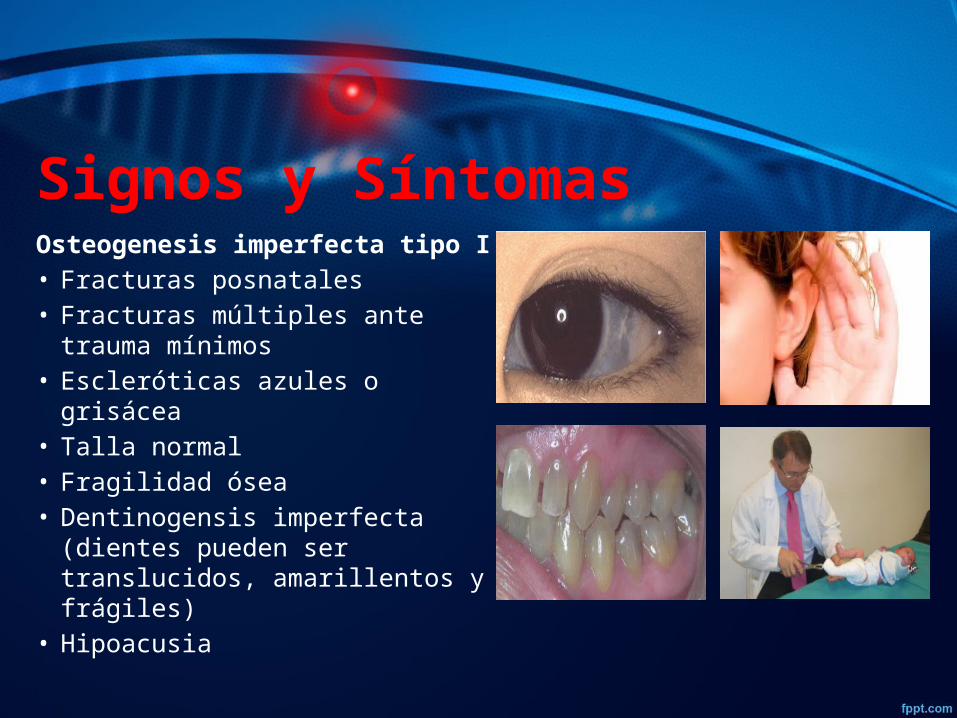
Signos y Síntomas Osteogenesis imperfecta tipo I:• Fracturas posnatales • Fracturas múltiples ante trauma
mínimos• Escleróticas azules o grisácea• Talla normal • Fragilidad ósea • Dentinogensis imperfecta (dientes
pueden ser translucidos, amarillentos y frágiles)
• Hipoacusia

Osteogenesis imperfecta tipo II:
• Fragilidad ósea extrema• Fragilidad del tejido
conjuntivo no óseo• Presencia de “islas” aisladas
de mineralización en el cráneo• Deformidad ósea • Escleróticas azules • Gran mortalidad perinatal y
fetal por fracturas intrauterina

Osteogenesis imperfecta tipo III:
• Retraso de crecimiento • Fracturas múltiples • Cifoescoliosis progresiva• Esclerótica azules al nacer • Hipoacusia • Dentinogenia imperfeta • Por lo general no son
ambulatorios

Osteogenesis imperfecta tipo IV:• Fracturas posnatales leves a
moderadas• Deformidades leves a
moderadas• Perdida prematura de la
audición • Escleróticas normales• Anormalidades dentales• Talla baja

Diagnostico
• Un examen físico • Se puede realizar una
densitometría• El diagnóstico prenatal de la
enfermedad: • Ultrasonido • Examen del tejido coriónico • Amniocentesis

Tratamiento No quirúrgico: - Terapia física- Útiles de soporte, como férulas, ortodoncia, etc. En los casos más graves, se tienen que emplear sillas de ruedas Farmacéutico: - Los bifosfonatos - Hormona de crecimiento
Quirúrgico: - Introducción de varillas intramedulares, conocidos como clavos quirúrgicos

Consejería Genética • Es necesario informar a ambos padres si ellos son
potencialmente portadores. Una vez diagnosticado el afectado, sus padres deben realizar un estudio para confirmar que ambos son portadores de la enfermedad.

¡GRACIAS!







